
Analysis of Fungal Microbiomes in Edible Medicinal Morindae Officinalis Radix and Alpiniae Oxyphyllae Fructus Using DNA Metabarcoding

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction
2.3. Polymerase Chain Reaction (PCR) Amplification and High-Throughput Sequencing (HTS)
2.4. Data Analysis
3. Results
3.1. Diversity Analysis of Fungal Microbiomes in MOR and AOF Samples
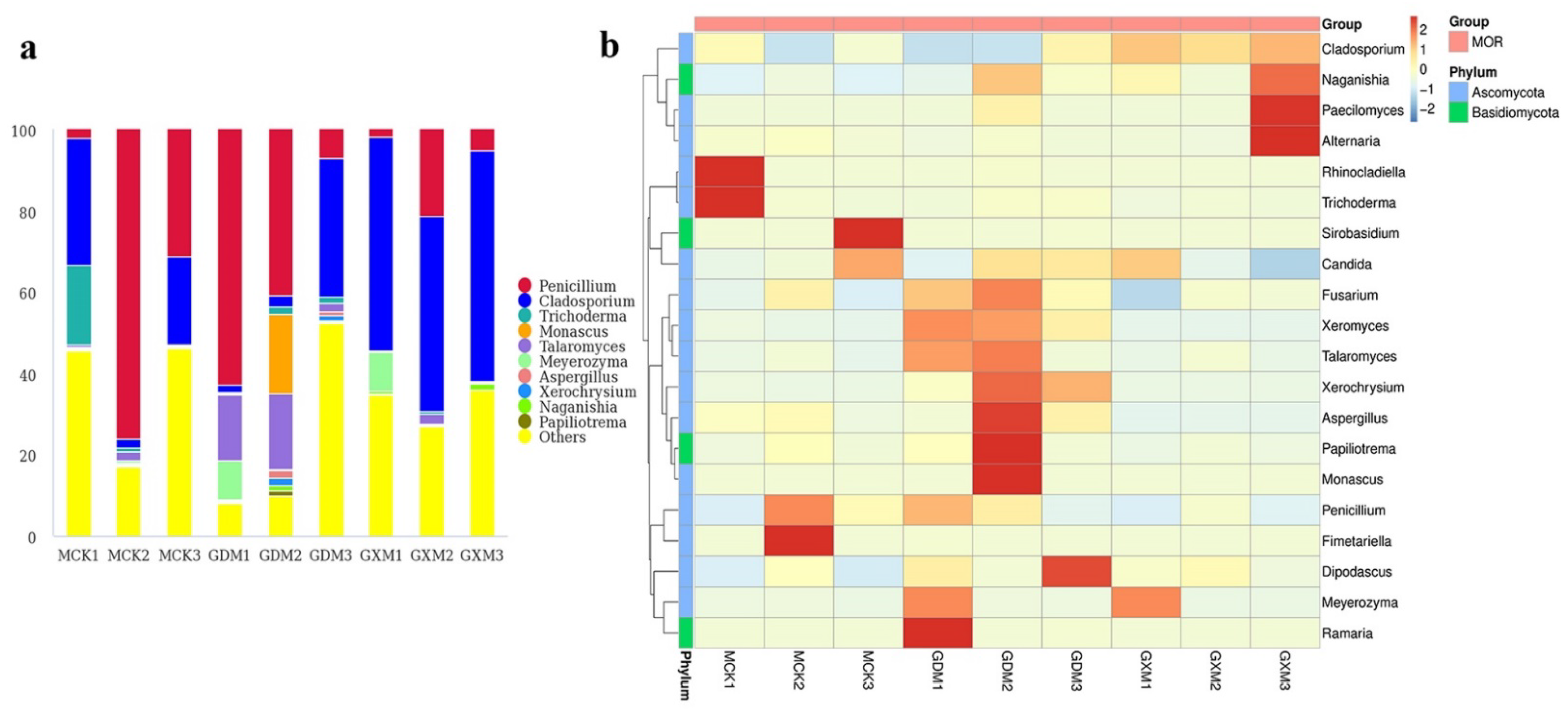
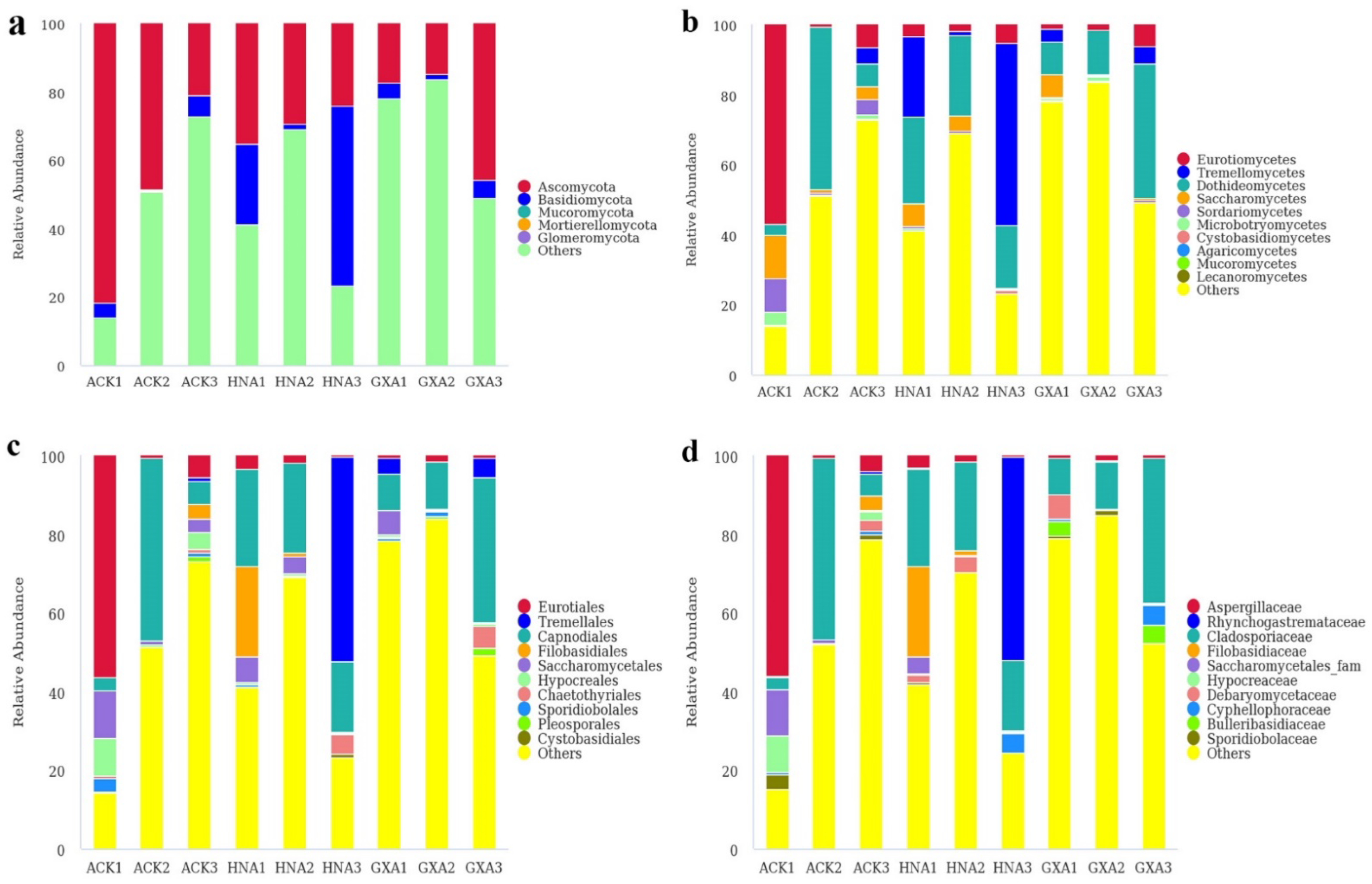
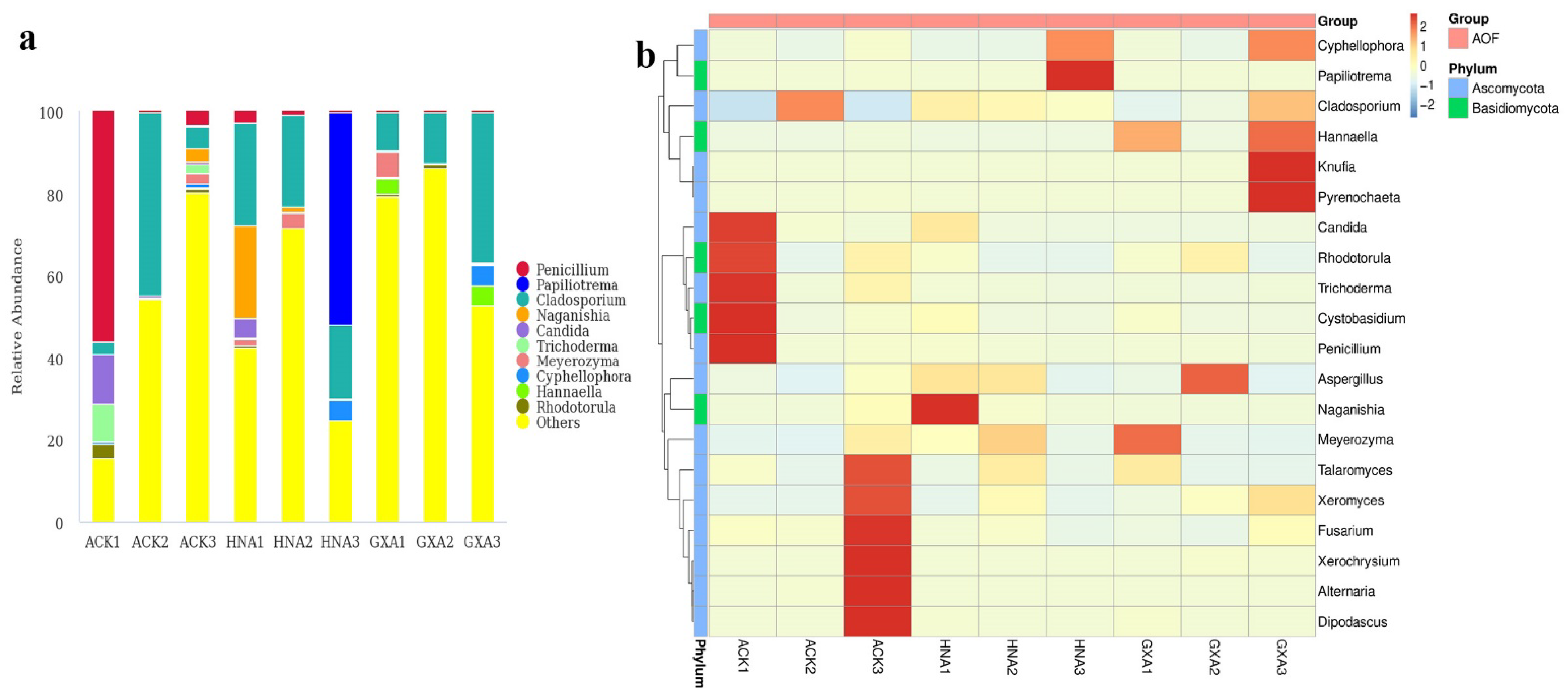
3.2. Composition of Fungal Microbiomes in MOR and AOF Samples
3.3. Comparison of Fungal Microbiomes in MOR and AOF
4. Discussion
4.1. The Necessity of Characterizing Fungal Microbiomes in MOR and AOF Samples
4.2. DNA Metabarcoding Is a Powerful Tool for the Analysis of Fungal Diversity in CHMs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, K. Some aspects of toxic contaminants in herbal medicines. Chemosphere 2003, 52, 1361–1371. [Google Scholar] [CrossRef] [Green Version]
- Ghisleni, D.D.M.; Braga, M.D.S.; Kikuchi, I.S.; Braşoveanu, M.; Nemţanu, M.R.; Dua, K.; Pinto, T.D.J.A. The Microbial Quality Aspects and Decontamination Approaches for the Herbal Medicinal Plants and Products: An in-Depth Review. Curr. Pharm. Des. 2016, 22, 4264–4287. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Lima, C.M.; Fujishima, M.A.T.; de Paula Lima, B.; Mastroianni, P.C.; de Sousa, F.F.O.; da Silva, J.O. Microbial contamination in herbal medicines: A serious health hazard to elderly consumers. BMC Complement. Med. Ther. 2020, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Keter, L.; Too, R.; Mwikwabe, N.; Mutai, C.; Orwa, J.; Mwamburi, L.; Ndwigah, S.; Bii, C.; Korir, R. Risk of Fungi Associated with Aflatoxin and Fumonisin in Medicinal Herbal Products in the Kenyan Market. Sci. World J. 2017, 2017, 1892972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinholds, I.; Bogdanova, E.; Pugajeva, I.; Alksne, L.; Stalberga, D.; Valcina, O.; Bartkevics, V. Determination of Fungi and Multi-Class Mycotoxins in Camelia sinensis and Herbal Teas and Dietary Exposure Assessment. Toxins 2020, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Waśkiewicz, A.; Beszterda, M.; Bocianowski, J.; Goliński, P. Natural occurrence of fumonisins and ochratoxin A in some herbs and spices commercialized in Poland analyzed by UPLC–MS/MS method. Food Microbiol. 2013, 36, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Hu, Y.; Gao, D.; Luo, Y.I.; Chen, A.J.; Jiao, X.; Gao, W. Occurrence of Toxigenic Fungi and Mycotoxins on Root Herbs from Chinese Markets. J. Food Prot. 2018, 81, 754–761. [Google Scholar] [CrossRef]
- Chinese Pharmacopeia Commission. Pharmacopoeia of People’s Republic of China (Part 1); China Medical Science Press: Beijing, China, 2020; pp. 83, 303. [Google Scholar]
- Yip, K.; Xu, J.; Zhou, S.; Lau, Y.; Chen, Q.; Tang, Y.; Yang, Z.; Yao, Z.; Ding, P.; Chen, H.; et al. Characterization of Chemical Component Variations in Different Growth Years and Tissues of Morindae Officinalis Radix by Integrating Metabolomics and Glycomics. J. Agric. Food Chem. 2019, 67, 7304–7314. [Google Scholar] [CrossRef]
- Liang, J.; Liang, J.; Hao, H.; Lin, H.; Wang, P.; Wu, Y.; Jiang, X.; Fu, C.; Li, Q.; Ding, P.; et al. The Extracts of Morinda officinalis and Its Hairy Roots Attenuate Dextran Sodium Sulfate-Induced Chronic Ulcerative Colitis in Mice by Regulating Inflammation and Lymphocyte Apoptosis. Front. Immunol. 2017, 8, 905. [Google Scholar] [CrossRef]
- Song, B.; Wang, F.; Wang, W. Effect of Aqueous Extract from Morinda officinalis F. C. How on Microwave-Induced Hypothalamic-Pituitary-Testis Axis Impairment in Male Sprague-Dawley Rats. Evid. Based Complement. Altern. Med. 2015, 2015, 360730. [Google Scholar] [CrossRef] [Green Version]
- Chi, L.; Khan, I.; Lin, Z.; Zhang, J.; Lee, M.Y.S.; Leong, W.; Hsiao, W.L.W.; Zheng, Y. Fructo-oligosaccharides from Morinda officinalis remodeled gut microbiota and alleviated depression features in a stress rat model. Phytomedicine 2020, 67, 153157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Q.; Yang, H.; Liu, W.; Zhang, N.; Qin, L.; Xin, H. Monotropein isolated from the roots of Morinda officinalis increases osteoblastic bone formation and prevents bone loss in ovariectomized mice. Fitoterapia 2016, 110, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Xin, H.L.; Xu, Y.M.; Shen, Y.; He, Y.Q.; Hsien-Yeh; Lin, B.; Song, H.T.; Yang, H.Y.; Qin, L.P.; et al. Morinda officinalis How. A comprehensive review of traditional uses, phytochemistry and pharmacology. J. Ethnopharmacol. 2018, 213, 230–255. [Google Scholar] [CrossRef] [PubMed]
- Qing, Z.J.; Yong, W.; Hui, L.Y.; Yong, L.W.; Long, L.H.; Ao, D.J.; Xia, P.L. Two new natural products from the fruits of Alpinia oxyphylla with inhibitory effects on nitric oxide production in lipopolysaccharide-activated RAW264.7 macrophage cells. Arch. Pharm. Res. 2012, 35, 2143–2146. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Chen, F.; Wang, J.F.; Wang, Y.; Zhang, J.Q.; Guo, T. Analysis of nine compounds from Alpinia oxyphylla fruit at different harvest time using UFLC-MS/MS and an extraction method optimized by orthogonal design. Chem. Cent. J. 2013, 7, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, L.J.; Guan, S.; Shi, G.F.; Bao, Y.M.; Duan, Y.L.; Jiang, B. Protocatechuic acid from Alpinia oxyphylla against MPP+-induced neurotoxicity in PC12 cells. Food Chem. Toxicol. 2006, 44, 436–443. [Google Scholar] [CrossRef]
- Shi, G.F.; An, L.J.; Jiang, B.; Guan, S.; Bao, Y.M. Alpinia protocatechuic acid protects against oxidative damage in vitro and reduces oxidative stress in vivo. Neurosci. Lett. 2006, 403, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Huang, D.; Zhang, D.; Wang, X.; Cao, H.; Zhang, Q.; Yan, C. Investigation of inulins from the roots of Morinda officinalis for potential therapeutic application as anti-osteoporosis agent. Int. J. Biol. Macromol. 2018, 120, 170–179. [Google Scholar] [CrossRef]
- Zhao, X.; Wei, J.; Zhou, Y.; Kong, W.; Yang, M. Quality evaluation of Alpinia oxyphylla after Aspergillus flavus infection for storage conditions optimization. AMB Express 2017, 7, 151. [Google Scholar] [CrossRef] [Green Version]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Christian, B.; Eske, W. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 2012, 21, 2045–2050. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Duan, N.; Xu, H.; Xie, T.N.; Xue, Y.R.; Liu, C.H. CTAB-PEG DNA Extraction from Fungi with High Contents of Polysaccharides. Mol. Biol. 2018, 52, 718–726. [Google Scholar] [CrossRef]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes—Application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and Direct Sequencing of Fungal Ribosomal Rna Genes for Phylogenetics. In PCR Protocols; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2021, 1, 10–12. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glockner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pẽa, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Guo, M.; Jiang, W.; Yang, M.; Pang, X. Assessment of the Microbiome and Potential Aflatoxin Associated with the Medicinal Herb Platycladus orientalis. Front Microbiol. 2020, 11, 582679. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Guo, M.; Yang, M.; Mantri, N.; Chen, X.; Pang, X. High-throughput analysis of fungal communities in Myristicae Semen. LWT Food Sci. Technol. 2020, 128, 109499. [Google Scholar] [CrossRef]
- Guo, M.; Jiang, W.; Yang, M.; Dou, X.; Pang, X. Characterizing fungal communities in medicinal and edible Cassiae Semen using high-throughput sequencing. Int. J. Food Microbiol. 2020, 319, 108496. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, K.; Habrowska-Gorczynska, D.E.; Piastowska-Ciesielska, A.W. Zearalenone as an endocrine disruptor in humans. Environ. Toxicol. Pharmacol. 2016, 48, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Barros, G.; Zanon, M.S.; Palazzini, J.M.; Haidukowski, M.; Pascale, M.; Chulze, S. Trichothecenes and zearalenone production by Fusarium equiseti and Fusarium semitectum species isolated from Argentinean soybean. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2012, 29, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Jiang, W.; Luo, J.; Yang, M.; Pang, X. Analysis of the Fungal Community in Ziziphi Spinosae Semen through High-Throughput Sequencing. Toxins 2018, 10, 494. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Qiu, F.; Kong, W.; Wei, J.; Yang, M.; Luo, Z.; Qin, J.; Ma, X. Co-occurrence of aflatoxin B1, B2, G1, G2 and ochrotoxin A in Glycyrrhiza uralensis analyzed by HPLC-MS/MS. Food Control 2013, 32, 216–221. [Google Scholar] [CrossRef]
- Prouillac, C.; Koraichi, F.; Videmann, B.; Mazallon, M.; Rodriguez, F.; Baltas, M.; Lecoeur, S. In vitro toxicological effects of estrogenic mycotoxins on human placental cells: Structure activity relationships. Toxicol. Appl. Pharmacol. 2012, 259, 366–375. [Google Scholar] [CrossRef]
- Zinedine, A.; Soriano, J.M.; Molto, J.C.; Manes, J. Review on the toxicity, occurrence, metabolism, detoxification, regulations and intake of zearalenone: An oestrogenic mycotoxin. Food Chem. Toxicol. 2007, 45, 1–18. [Google Scholar] [CrossRef]
- Kong, W.; Wei, R.; Logrieco, A.F.; Wei, J.; Wen, J.; Xiao, X.; Yang, M. Occurrence of toxigenic fungi and determination of mycotoxins by HPLC-FLD in functional foods and spices in China markets. Food Chem. 2014, 146, 320–326. [Google Scholar] [CrossRef]
- Nian, Y.; Wang, H.; Ying, G.; Yang, M.; Wang, Z.; Kong, W.; Yang, S. Transfer rates of aflatoxins from herbal medicines to decoctions determined by an optimized high-performance liquid chromatography with fluorescence detection method. J. Pharm. Pharmacol. 2018, 70, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Shim, W.B.; Ha, K.S.; Kim, M.G.; Kim, J.S.; Chung, D.H. Evaluation of the transfer rate of ochratoxin a to decoctions of herbal medicines. Food Sci. Biotechnol. 2014, 23, 2103–2108. [Google Scholar] [CrossRef]
- Xia, F.; Chen, X.; Guo, M.Y.; Bai, X.H.; Liu, Y.; Shen, G.R.; Li, Y.L.; Lin, J.; Zhou, X.W. High-throughput sequencing-based analysis of endogenetic fungal communities inhabiting the Chinese Cordyceps reveals unexpectedly high fungal diversity. Sci. Rep. 2016, 6, 33437. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Li, H.; Jiang, D.; Meng, J.; Zhang, F.; Xu, Q.; Chen, X.; Liu, C.; Yang, Y. Analysis of Fungi on Coix (Coix lacryma-jobi) Seed and the Effect of Its Aqueous Extract on the Growth of Aspergillus flavus. J. Food Prot. 2019, 82, 1775–1782. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sample ID | Group | Moldy | Group | Source | GenBank Accession No. |
|---|---|---|---|---|---|---|
| Morindae Officinalis Radix | MCK1 | MOR | No | NM | Haikou, Hainan | SAMN19591296 |
| Morindae Officinalis Radix | MCK2 | MOR | No | NM | Haikou, Hainan | SAMN19591297 |
| Morindae Officinalis Radix | MCK3 | MOR | No | NM | Haikou, Hainan | SAMN19591298 |
| Morindae Officinalis Radix | GDM1 | MOR | Yes | MM | Qingping, Guangdong | SAMN19591299 |
| Morindae Officinalis Radix | GDM2 | MOR | Yes | MM | Qingping, Guangdong | SAMN19591300 |
| Morindae Officinalis Radix | GDM3 | MOR | Yes | MM | Qingping, Guangdong | SAMN19591301 |
| Morindae Officinalis Radix | GXM1 | MOR | Yes | MM | Yulin, Guangxi | SAMN19591302 |
| Morindae Officinalis Radix | GXM2 | MOR | Yes | MM | Yulin, Guangxi | SAMN19591303 |
| Morindae Officinalis Radix | GXM3 | MOR | Yes | MM | Yulin, Guangxi | SAMN19591304 |
| Alpiniae Oxyphyllae Fructus | ACK1 | AOF | No | NA | Haikou, Hainan | SAMN19591305 |
| Alpiniae Oxyphyllae Fructus | ACK2 | AOF | No | NA | Haikou, Hainan | SAMN19591306 |
| Alpiniae Oxyphyllae Fructus | ACK3 | AOF | No | NA | Haikou, Hainan | SAMN19591307 |
| Alpiniae Oxyphyllae Fructus | HNA1 | AOF | Yes | MA | Qingping, Guangdong | SAMN19591308 |
| Alpiniae Oxyphyllae Fructus | HNA2 | AOF | Yes | MA | Qingping, Guangdong | SAMN19591309 |
| Alpiniae Oxyphyllae Fructus | HNA3 | AOF | Yes | MA | Qingping, Guangdong | SAMN19591310 |
| Alpiniae Oxyphyllae Fructus | GXA1 | AOF | Yes | MA | Yulin, Guangxi | SAMN19591311 |
| Alpiniae Oxyphyllae Fructus | GXA2 | AOF | Yes | MA | Yulin, Guangxi | SAMN19591312 |
| Alpiniae Oxyphyllae Fructus | GXA3 | AOF | Yes | MA | Yulin, Guangxi | SAMN19591313 |
| Sample No. | Shannon | Simpson | Chao1 | Ace | Goods_Coverage |
|---|---|---|---|---|---|
| MCK1 | 4.055 | 0.857 | 249.794 | 257.651 | 0.999 |
| MCK2 | 3.791 | 0.779 | 364.441 | 354.497 | 0.998 |
| MCK3 | 3.971 | 0.865 | 269.150 | 282.910 | 0.999 |
| GDM1 | 4.412 | 0.910 | 315.250 | 297.879 | 0.998 |
| GDM2 | 4.618 | 0.902 | 321.588 | 323.193 | 0.999 |
| GDM3 | 4.068 | 0.840 | 347.370 | 333.659 | 0.998 |
| GXM1 | 3.269 | 0.721 | 274.000 | 280.869 | 0.998 |
| GXM2 | 3.441 | 0.751 | 243.077 | 253.748 | 0.999 |
| GXM3 | 3.098 | 0.679 | 280.278 | 294.317 | 0.998 |
| ACK1 | 3.122 | 0.740 | 349.241 | 330.054 | 0.998 |
| ACK2 | 3.403 | 0.766 | 290.250 | 300.323 | 0.998 |
| ACK3 | 4.050 | 0.745 | 424.241 | 402.027 | 0.998 |
| HNA1 | 3.706 | 0.843 | 294.286 | 309.556 | 0.998 |
| HNA2 | 4.886 | 0.931 | 315.038 | 308.207 | 0.999 |
| HNA3 | 3.786 | 0.803 | 273.933 | 276.385 | 0.999 |
| GXA1 | 3.476 | 0.712 | 311.838 | 322.078 | 0.998 |
| GXA2 | 3.398 | 0.761 | 238.037 | 244.68 | 0.999 |
| GXA3 | 4.064 | 0.839 | 257.500 | 269.869 | 0.999 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Chen, X.; Guo, M.; Yu, J.; Yang, M.; Pang, X. Analysis of Fungal Microbiomes in Edible Medicinal Morindae Officinalis Radix and Alpiniae Oxyphyllae Fructus Using DNA Metabarcoding. Foods 2022, 11, 1748. https://doi.org/10.3390/foods11121748
Jiang W, Chen X, Guo M, Yu J, Yang M, Pang X. Analysis of Fungal Microbiomes in Edible Medicinal Morindae Officinalis Radix and Alpiniae Oxyphyllae Fructus Using DNA Metabarcoding. Foods. 2022; 11(12):1748. https://doi.org/10.3390/foods11121748
Chicago/Turabian StyleJiang, Wenjun, Xuyu Chen, Mengyue Guo, Jingsheng Yu, Meihua Yang, and Xiaohui Pang. 2022. "Analysis of Fungal Microbiomes in Edible Medicinal Morindae Officinalis Radix and Alpiniae Oxyphyllae Fructus Using DNA Metabarcoding" Foods 11, no. 12: 1748. https://doi.org/10.3390/foods11121748