

Coupling Hydrophilic Interaction Chromatography and Reverse-Phase Chromatography for Improved Direct Analysis of Grape Seed Proanthocyanidins
 ,
, 
Abstract
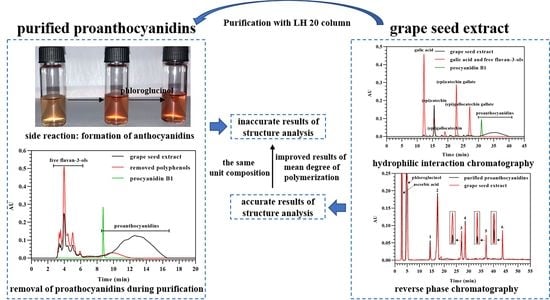
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Purification of Proanthocyanidins from Grape Seed Extract with a Sephadex LH-20 Column
2.3. Depolymerization of Proanthocyanidins in the Presence of Excess Phloroglucinol
2.4. Optical Absorption Analysis
2.5. Analysis of Cleavage Products by RPHPLC-MS
2.6. Analysis of Free Flavan-3-Ols in Grape Seed Extract by HILIC
2.7. Analysis of Free Flavan-3-Ols and Proanthocyanidins Removed from Grape Seed Extract by HILIC
2.8. Analysis of Molecular Weight Distribution by Gel Permeation Chromatography (GPC)
2.9. Determination of Proanthocyanidins with Butanol-HCl Assay
2.10. Statistical Analysis
3. Results and Discussion
3.1. Transformation of Proanthocyanidins to Anthocyanidins along with Acid-Catalyzed Depolymerization
3.2. The Effect of Cyanidin Formation on the Molecular Weight Distribution of Proanthocyanidins
3.3. Purification of Proanthocyanidins from Grape Seed Extract
3.4. Analysis of Flavan-3-Ol Monomers in Grape Seed Extract
3.5. Direct Analysis of Proanthocyanidins in Grape Seed Extract in the Presence of Excess Phloroglucinol
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rasmussen, S.E.; Frederiksen, H.; Struntze Krogholm, K.; Poulsen, L. Dietary proanthocyanidins: Occurrence, dietary intake, bioavailability, and protection against cardiovascular disease. Mol. Nutr. Food Res. 2005, 49, 159–174. [Google Scholar] [CrossRef]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Beecher, G.; Holden, J.; Haytowitz, D.; Gebhardt, S.; Prior, R.L. Concentrations of proanthocyanidins in common foods and estimations of normal consumption. J. Nutr. 2004, 134, 613–617. [Google Scholar] [PubMed]
- Rauf, A.; Imran, M.; Abu-Izneid, T.; Iahtisham Ul, H.; Patel, S.; Pan, X.; Naz, S.; Sanches Silva, A.; Saeed, F.; Rasul Suleria, H.A. Proanthocyanidins: A comprehensive review. Biomed. Pharmacother. 2019, 116, 108999. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Zhang, Y.; Shen, X.; Cao, Y.; Shi, J.; Ye, X.; Chen, S. Rethinking the mechanism of the health benefits of proanthocyanidins: Absorption, metabolism, and interaction with gut microbiota. Compr. Rev. Food Sci. Food Saf. 2019, 18, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Kalili, K.M.; de Villiers, A. Off-line comprehensive 2-dimensional hydrophilic interaction×reversed phase liquid chromatography analysis of procyanidins. J. Chromatogr. A 2009, 1216, 6274–6284. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Jones, G.P. Analysis of proanthocyanidin cleavage products following acid-catalysis in the presence of excess phloroglucinol. J. Agric. Food Chem. 2001, 49, 1740–1746. [Google Scholar] [CrossRef]
- Wang, Y.; Chung, S.-J.; Song, W.O.; Chun, O.K. Estimation of daily proanthocyanidin intake and major food sources in the US diet. J. Nutr. 2011, 141, 447–452. [Google Scholar] [CrossRef]
- Yanagida, A.; Murao, H.; Ohnishi-Kameyama, M.; Yamakawa, Y.; Shoji, A.; Tagashira, M.; Kanda, T.; Shindo, H.; Shibusawa, Y. Retention behavior of oligomeric proanthocyanidins in hydrophilic interaction chromatography. J. Chromatogr. A 2007, 1143, 153–161. [Google Scholar] [CrossRef]
- Ou, K.; Gu, L. Absorption and metabolism of proanthocyanidins. J. Funct. Foods 2014, 7, 43–53. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Waterhouse, A.L. Analysis of pigmented high-molecular-mass grape phenolics using ion-pair, normal-phase high-performance liquid chromatography. J. Chromatogr. A 2000, 866, 25–34. [Google Scholar] [CrossRef]
- Bartzoka, E.D.; Lange, H.; Mosesso, P.; Crestini, C. Synthesis of nano- and microstructures from proanthocyanidins, tannic acid and epigallocatechin-3-O-gallate for active delivery. Green Chem. 2017, 19, 5074–5091. [Google Scholar] [CrossRef]
- Hollands, W.J.; Voorspoels, S.; Jacobs, G.; Aaby, K.; Meisland, A.; Garcia-Villalba, R.; Tomas-Barberan, F.; Piskula, M.K.; Mawson, D.; Vovk, I.; et al. Development, validation and evaluation of an analytical method for the determination of monomeric and oligomeric procyanidins in apple extracts. J. Chromatogr. A 2017, 1495, 46–56. [Google Scholar] [CrossRef]
- Matthews, S.; Mila, I.; Scalbert, A.; Pollet, B.; Lapierre, C.; Hervé du Penhoat, C.L.M.; Rolando, C.; Donnelly, D.M.X. Method for estimation of proanthocyanidins based on their acid depolymerization in the presence of nucleophiles. J. Agric. Food Chem. 1997, 45, 1195–1201. [Google Scholar] [CrossRef]
- Luo, L.; Cui, Y.; Cheng, J.; Fang, B.; Wei, Z.; Sun, B. An approach for degradation of grape seed and skin proanthocyanidin polymers into oligomers by sulphurous acid. Food Chem. 2018, 256, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Shay, P.-E.; Trofymow, J.A.; Constabel, C.P. An improved butanol-HCl assay for quantification of water-soluble, acetone:methanol-soluble, and insoluble proanthocyanidins (condensed tannins). Plant Methods 2017, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Georgiev, V.; Ananga, A.; Tsolova, V. Recent advances and uses of grape flavonoids as nutraceuticals. Nutrients 2014, 6, 391–415. [Google Scholar] [CrossRef]
- Graf, B.L.; Raskin, I.; Cefalu, W.T.; Ribnicky, D.M. Plant-derived therapeutics for the treatment of metabolic syndrome. Curr. Opin. Investig. Drugs 2010, 11, 1107–1115. [Google Scholar]
- Nandakumar, V.; Singh, T.; Katiyar, S.K. Multi-targeted prevention and therapy of cancer by proanthocyanidins. Cancer Lett. 2008, 269, 378–387. [Google Scholar] [CrossRef]
- Yang, H.; Ye, X.; Liu, D.; Chen, J.; Zhang, J.; Shen, Y.; Yu, D. Characterization of unusual proanthocyanidins in leaves of bayberry (Myrica rubra Sieb. et Zucc.). J. Agric. Food Chem. 2011, 59, 1622–1629. [Google Scholar] [CrossRef]
- Bartzoka, E.D.; Lange, H.; Thiel, K.; Crestini, C. Coordination complexes and one-step assembly of lignin for versatile nanocapsule engineering. ACS Sustain. Chem. Eng. 2016, 4, 5194–5203. [Google Scholar] [CrossRef]
- Grabber, J.H.; Zeller, W.E.; Mueller-Harvey, I. Acetone enhances the direct analysis of procyanidin-and prodelphinidin-based condensed tannins in Lotus species by the butanol-HCl-iron assay. J. Agric. Food Chem. 2013, 61, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Porter, L.J.; Hrstich, L.N.; Chan, B.G. The conversion of procyanidins and prodelphinidins to cyanidin and delphinidin. Phytochemistry 1985, 25, 223–230. [Google Scholar] [CrossRef]
- Zhang, Z.; Kou, X.; Fugal, K.; McLaughlin, J. Comparison of HPLC methods for determination of anthocyanins and anthocyanidins in bilberry extracts. J. Agric. Food Chem. 2004, 52, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Ştefănuţ, M.N.; Căta, A.; Pop, R.; Moşoarcă, C.; Zamfir, A.D. Anthocyanins HPLC-DAD and MS characterization, total phenolics, and antioxidant activity of some berries extracts. Anal. Lett. 2011, 44, 2843–2855. [Google Scholar] [CrossRef]
- Liu, H.; Zou, T.; Gao, J.M.; Gu, L. Depolymerization of cranberry procyanidins using (+)-catechin, (-)-epicatechin, and (-)-epigallocatechin gallate as chain breakers. Food Chem. 2013, 141, 488–494. [Google Scholar] [CrossRef]
- Gong, Y.; Pegg, R.B. Separation of ellagitannin-rich phenolics from US pecans and Chinese hickory nuts using fused-core HPLC columns and their characterization. J. Agric. Food Chem. 2017, 65, 5810–5820. [Google Scholar] [CrossRef]
- Sommella, E.; Pepe, G.; Pagano, F.; Ostacolo, C.; Tenore, G.C.; Russo, M.T.; Novellino, E.; Manfra, M.; Campiglia, P. Detailed polyphenolic profiling of Annurca apple (M. pumila Miller cv Annurca) by a combination of RP-UHPLC and HILIC, both hyphenated to IT-TOF mass spectrometry. Food Res. Int. 2015, 76, 466–477. [Google Scholar] [CrossRef]
- Montero, L.; Herrero, M.; Prodanov, M.; Ibáñez, E.; Cifuentes, A. Characterization of grape seed procyanidins by comprehensive two-dimensional hydrophilic interaction × reversed phase liquid chromatography coupled to diode array detection and tandem mass spectrometry. Anal. Bioana. Chem. 2013, 405, 4627–4638. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, R.; Wang, Y.; Cheng, H.; Chen, S.; Ye, X.; Pan, H. Coupling Hydrophilic Interaction Chromatography and Reverse-Phase Chromatography for Improved Direct Analysis of Grape Seed Proanthocyanidins. Foods 2023, 12, 1319. https://doi.org/10.3390/foods12061319
Lin R, Wang Y, Cheng H, Chen S, Ye X, Pan H. Coupling Hydrophilic Interaction Chromatography and Reverse-Phase Chromatography for Improved Direct Analysis of Grape Seed Proanthocyanidins. Foods. 2023; 12(6):1319. https://doi.org/10.3390/foods12061319
Chicago/Turabian StyleLin, Ruge, Yi Wang, Huan Cheng, Shiguo Chen, Xingqian Ye, and Haibo Pan. 2023. "Coupling Hydrophilic Interaction Chromatography and Reverse-Phase Chromatography for Improved Direct Analysis of Grape Seed Proanthocyanidins" Foods 12, no. 6: 1319. https://doi.org/10.3390/foods12061319
APA StyleLin, R., Wang, Y., Cheng, H., Chen, S., Ye, X., & Pan, H. (2023). Coupling Hydrophilic Interaction Chromatography and Reverse-Phase Chromatography for Improved Direct Analysis of Grape Seed Proanthocyanidins. Foods, 12(6), 1319. https://doi.org/10.3390/foods12061319