
Red Cabbage Modulates Composition and Co-Occurrence Networks of Gut Microbiota in a Rodent Diet-Induced Obesity Model
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Establishment of Mice Obesity Model
2.2. Measurement of Physiological Indicators in Mice Samples
2.3. Fecal DNA Extraction and Quantitative PCR Analysis
2.4. Cecum DNA Extraction and 16S rRNA Sequencing
2.5. Sequence Data Analysis
2.6. Statistical Analysis and Visualization
3. Results
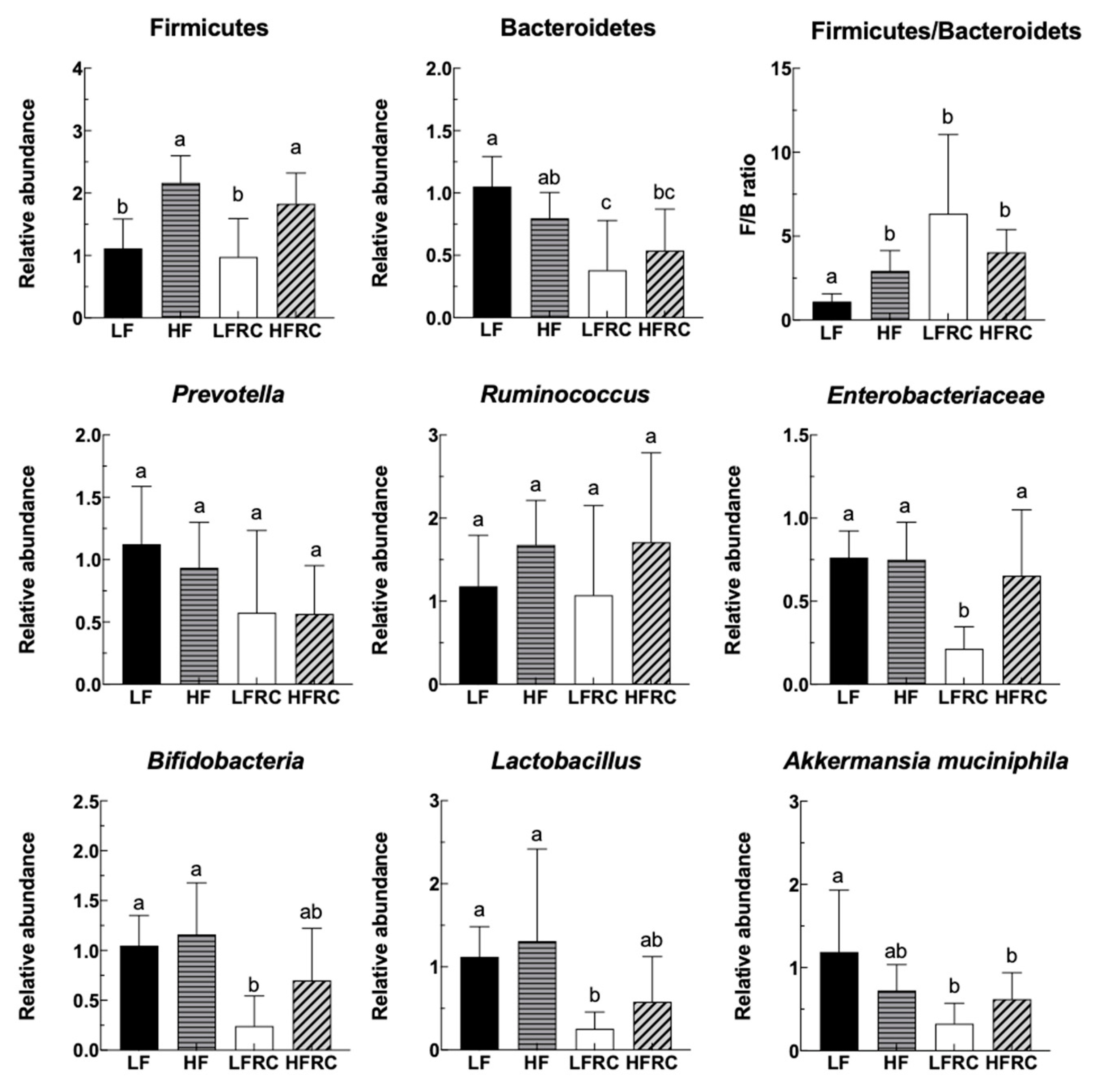
3.1. Effect of Consuming RC for 24 h on the Fecal Microbiota of Mice
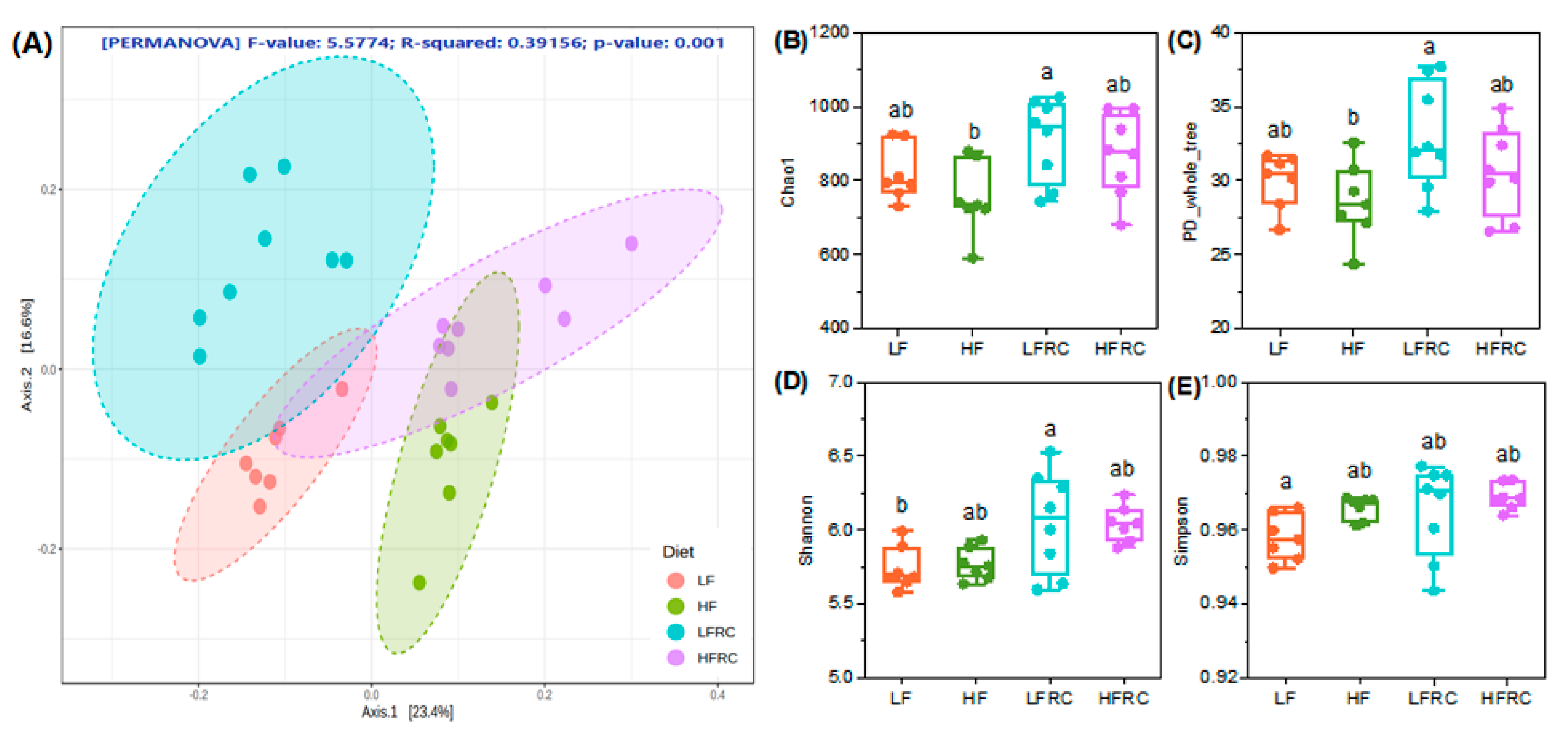
3.2. Effect of Consuming RC for 8 Weeks on Microbial Diversity in the Mice Cecum
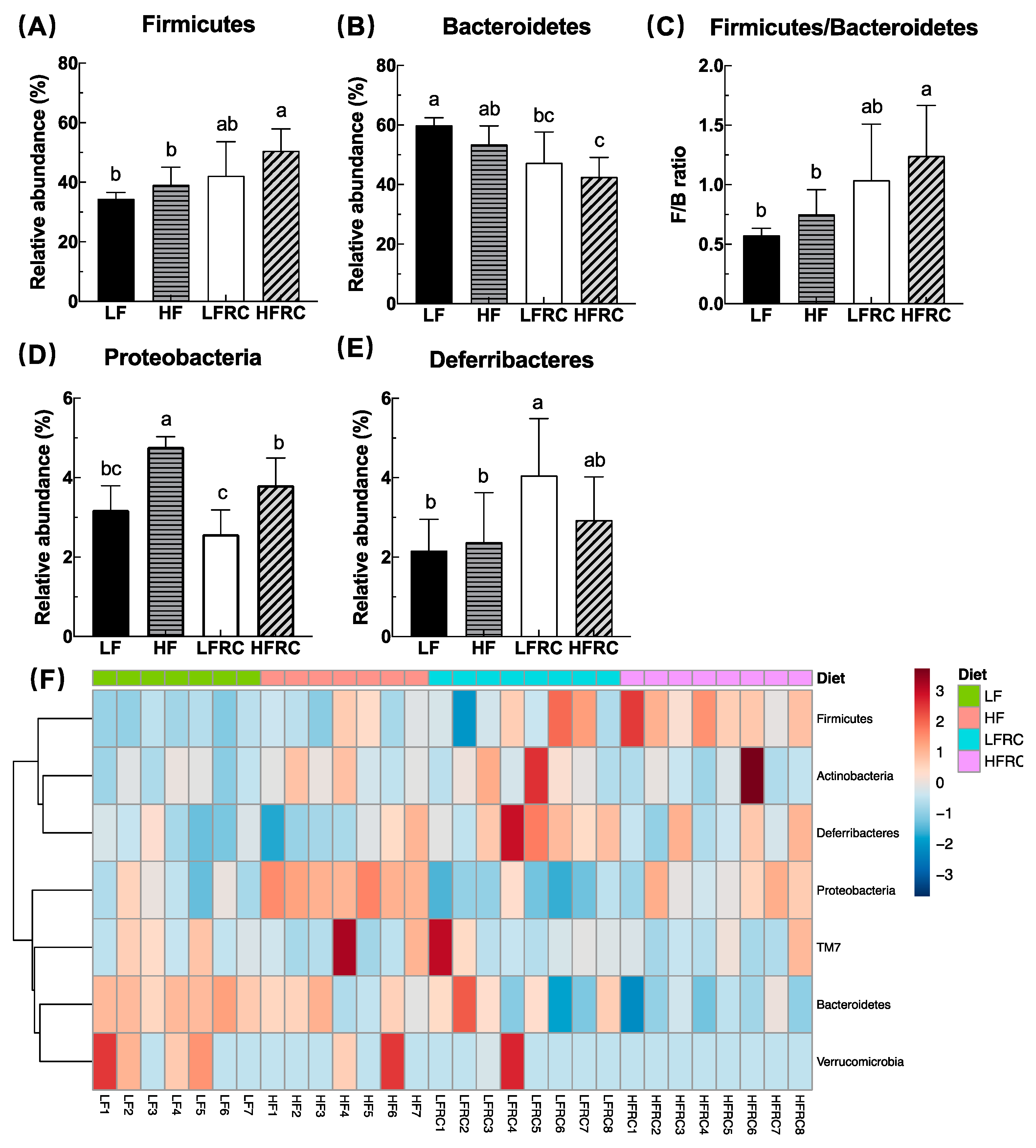
3.3. Effect of Consuming RC for 8 Weeks on the Composition of Cecal Microbiota in Mice
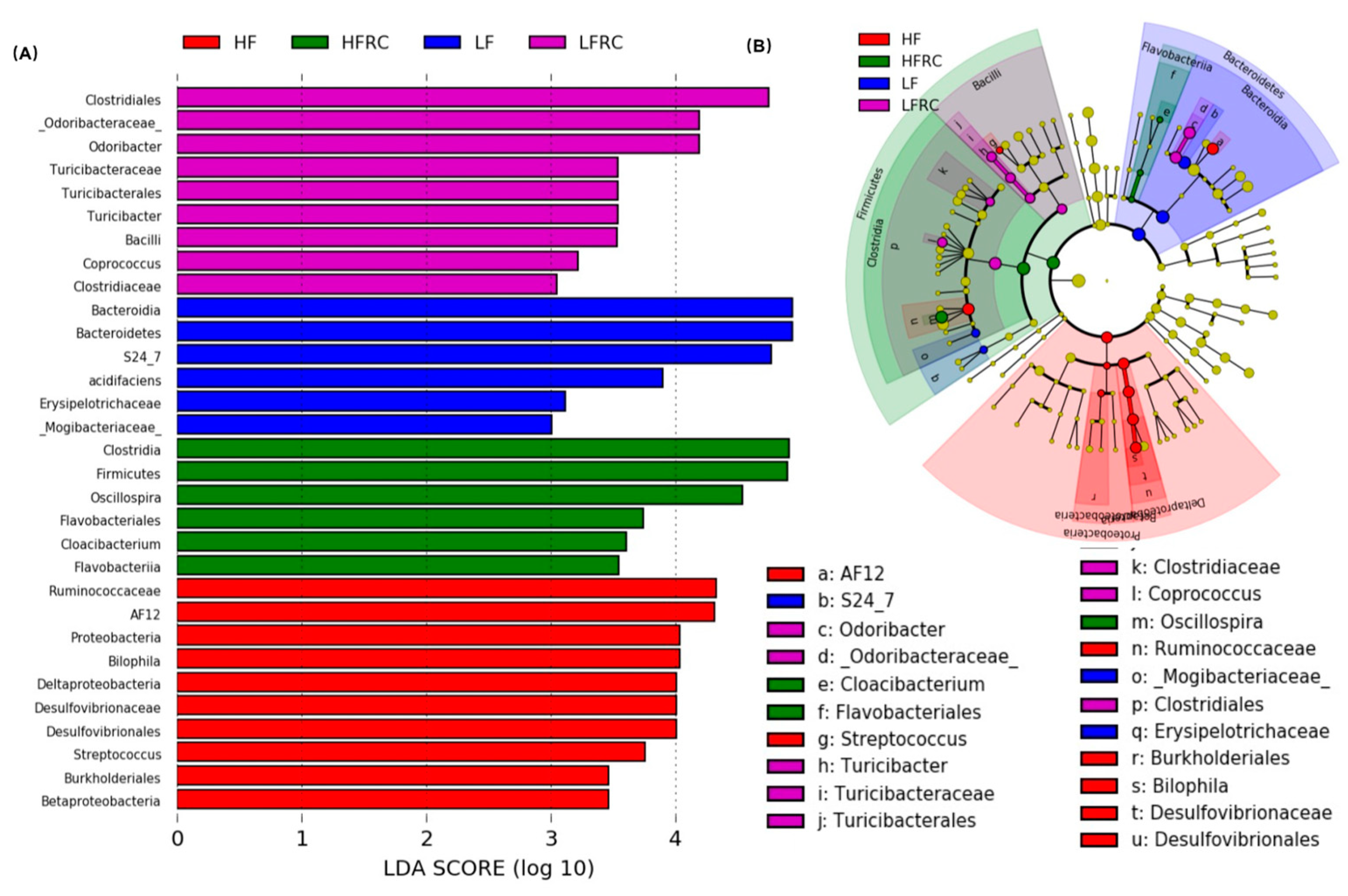
3.4. Identification of Microbial Biomarkers Associated with a Diet
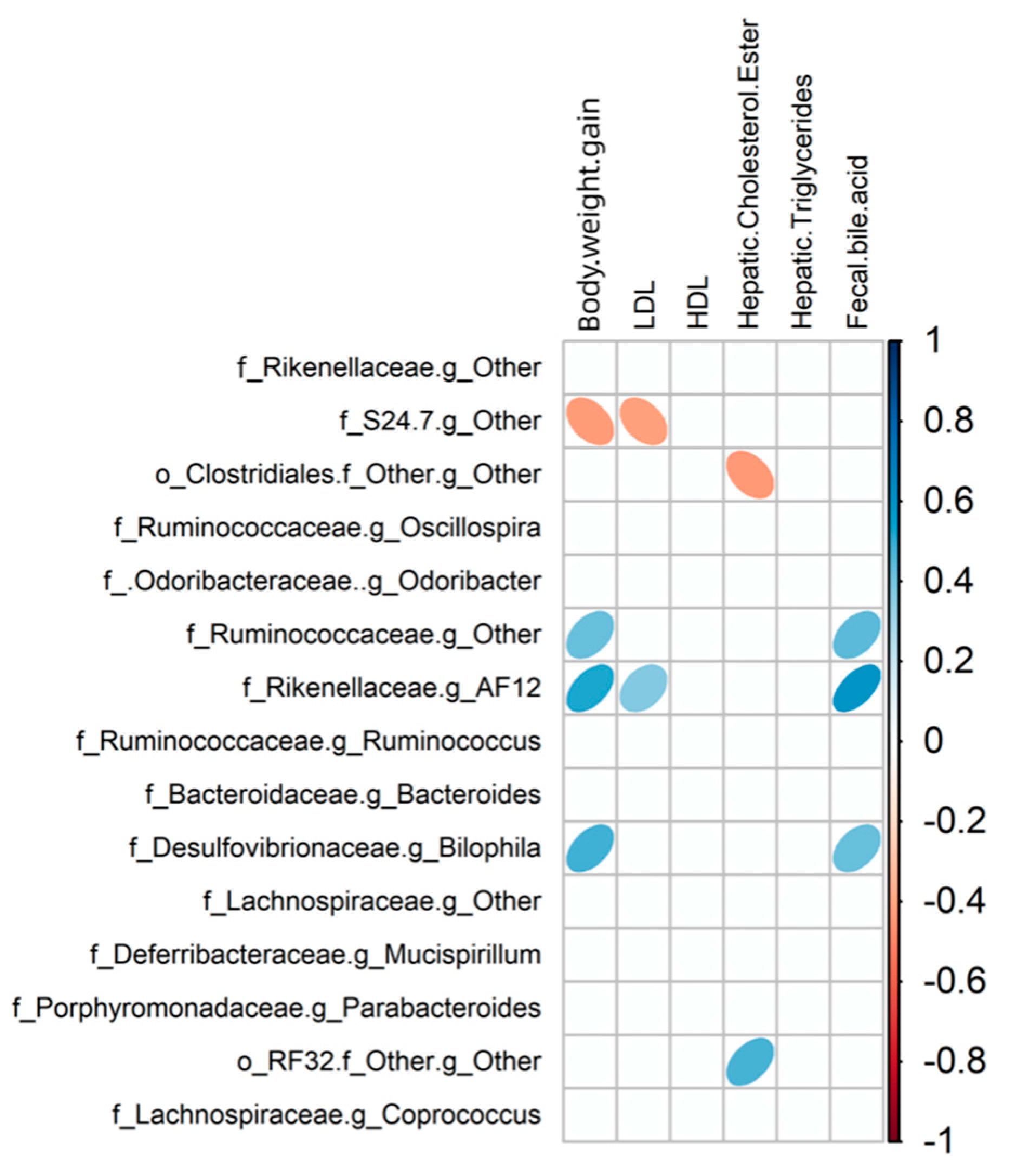
3.5. Relationship between Gut Microbiota Communities and Biochemical Indices in Mice
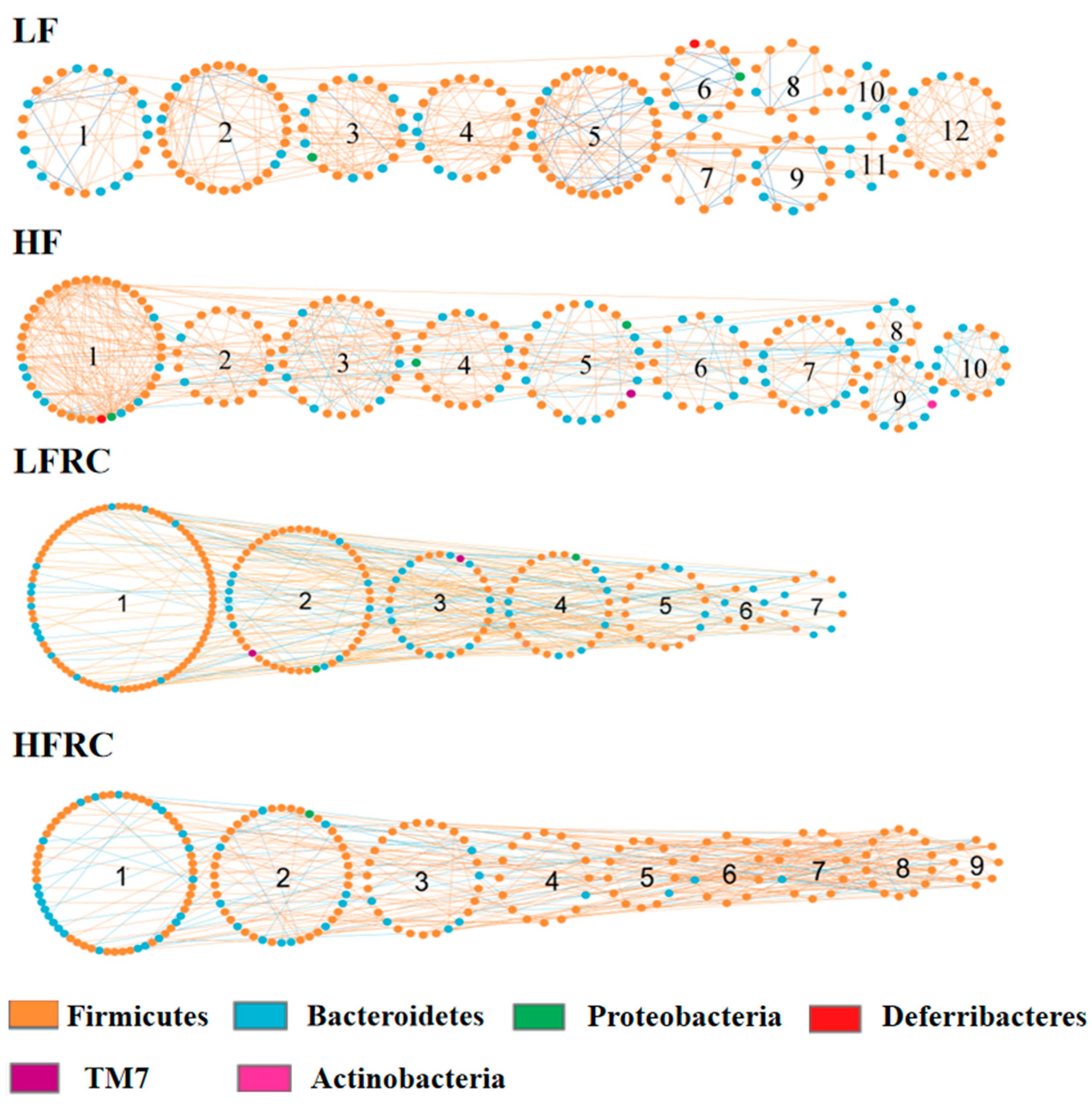
3.6. Effect of RC on the Gut Microbial Ecological Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: A review. Antonie Van Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. The gut microbiota and obesity: From correlation to causality. Nat. Rev. Microbiol. 2013, 11, 639–647. [Google Scholar] [CrossRef]
- Kelly, T.; Yang, W.; Chen, C.-S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. 2008, 32, 1431–1437. [Google Scholar] [CrossRef]
- Després, J.-P. Health consequences of visceral obesity. Ann. Med. 2001, 33, 534–541. [Google Scholar] [CrossRef]
- Kopelman, P. Health risks associated with overweight and obesity. Obes. Rev. 2007, 8, 13–17. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; Nilaweera, K.; Ross, P.R.; Shanahan, F.; O’Toole, P.W.; Cotter, P.D. The gut microbiota and its relationship to diet and obesity: New insights. Gut Microbes 2012, 3, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Martinez, K.B.; Leone, V.; Chang, E.B. Western diets, gut dysbiosis, and metabolic diseases: Are they linked? Gut Microbes 2017, 8, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef]
- Laparra, J.M.; Sanz, Y. Interactions of gut microbiota with functional food components and nutraceuticals. Pharmacol. Res. 2010, 61, 219–225. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559. [Google Scholar] [CrossRef]
- Zayed, A.; Sheashea, M.; Kassem, I.A.A.; Farag, M.A. Red and white cabbages: An updated comparative review of bioactives, extraction methods, processing practices, and health benefits. Crit. Rev. Food Sci. Nutr. 2023, 63, 7025–7042. [Google Scholar] [CrossRef]
- Podsędek, A.; Sosnowska, D.; Redzynia, M.; Anders, B. Antioxidant capacity and content of Brassica oleracea dietary antioxidants. Int. J. Food Sci. Technol. 2006, 41, 49–58. [Google Scholar] [CrossRef]
- Aravind, S.M.; Wichienchot, S.; Tsao, R.; Ramakrishnan, S.; Chakkaravarthi, S. Role of dietary polyphenols on gut microbiota, their metabolites and health benefits. Food Res. Int. 2021, 142, 110189. [Google Scholar] [CrossRef]
- Mithen, R.F.; Dekker, M.; Verkerk, R.; Rabot, S.; Johnson, I.T. The nutritional significance, biosynthesis and bioavailability of glucosinolates in human foods. J. Sci. Food Agric. 2000, 80, 967–984. [Google Scholar] [CrossRef]
- Kahalehili, H.M.; Newman, N.K.; Pennington, J.M.; Kolluri, S.K.; Kerkvliet, N.I.; Shulzhenko, N.; Morgun, A.; Ehrlich, A.K. Dietary indole-3-carbinol activates AhR in the gut, alters Th17-microbe interactions, and exacerbates insulitis in NOD mice. Front. Immunol. 2021, 11, 606441. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Busbee, P.B.; Menzel, L.; Alrafas, H.R.; Dopkins, N.; Becker, W.; Miranda, K.; Tang, C.; Chatterjee, S.; Singh, U.P.; Nagarkatti, M. Indole-3-carbinol prevents colitis and associated microbial dysbiosis in an IL-22–dependent manner. JCI Insight 2020, 5, e127551. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, F.; Paredes-Gonzalez, X.; Kong, A.-N.T. Dietary glucosinolates sulforaphane, phenethyl isothiocyanate, indole-3-carbinol/3, 3′-diindolylmethane: Antioxidative stress/inflammation, Nrf2, epigenetics/epigenomics and in vivo cancer chemopreventive efficacy. Curr. Pharmacol. Rep. 2015, 1, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jiang, X.; Xiao, Z.; Yu, L.; Pham, Q.; Sun, J.; Chen, P.; Yokoyama, W.; Yu, L.L.; Luo, Y.S. Red cabbage microgreens lower circulating low-density lipoprotein (LDL), liver cholesterol, and inflammatory cytokines in mice fed a high-fat diet. J. Agric. Food Chem. 2016, 64, 9161–9171. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, R.W.; Huang, H.; Fletcher, A.; Yu, L.; Pham, Q.; Yu, L.; He, Q.; Wang, T.T.Y. Inhibition of Tumor Growth by Dietary Indole-3-Carbinol in a Prostate Cancer Xenograft Model May Be Associated with Disrupted Gut Microbial Interactions. Nutrients 2019, 11, 467. [Google Scholar] [CrossRef] [PubMed]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Li, R.W.; Wu, S.; Li, W.; Huang, Y.; Gasbarre, L.C. Metagenome plasticity of the bovine abomasal microbiota in immune animals in response to Ostertagia ostertagi infection. PLoS ONE 2011, 6, e24417. [Google Scholar] [CrossRef]
- Li, R.W.; Wu, S.; Li, W.; Navarro, K.; Couch, R.D.; Hill, D.; Urban, J.F. Alterations in the porcine colon microbiota induced by the gastrointestinal nematode Trichuris suis. Infect. Immun. 2012, 80, 2150–2157. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Vendrame, S.; Guglielmetti, S.; Riso, P.; Arioli, S.; Klimis-Zacas, D.; Porrini, M. Six-week consumption of a wild blueberry powder drink increases bifidobacteria in the human gut. J. Agric. Food Chem. 2011, 59, 12815–12820. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Amaretti, A.; Leonardi, A.; Raimondi, S.; Simone, M.; Quartieri, A. Potential impact of probiotic consumption on the bioactivity of dietary phytochemicals. J. Agric. Food Chem. 2013, 61, 9551–9558. [Google Scholar] [CrossRef] [PubMed]
- Klinder, A.; Shen, Q.; Heppel, S.; Lovegrove, J.A.; Rowland, I.; Tuohy, K.M. Impact of increasing fruit and vegetables and flavonoid intake on the human gut microbiota. Food Funct. 2016, 7, 1788–1796. [Google Scholar] [CrossRef]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A common factor in human diseases. Biomed Res. Int. 2017, 2017, 9351507. [Google Scholar] [CrossRef]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum/Class; Order; Family | Genus | Diet | |||
|---|---|---|---|---|---|
| LF | HF | LFRC | HFRC | ||
| Bacteroidetes | |||||
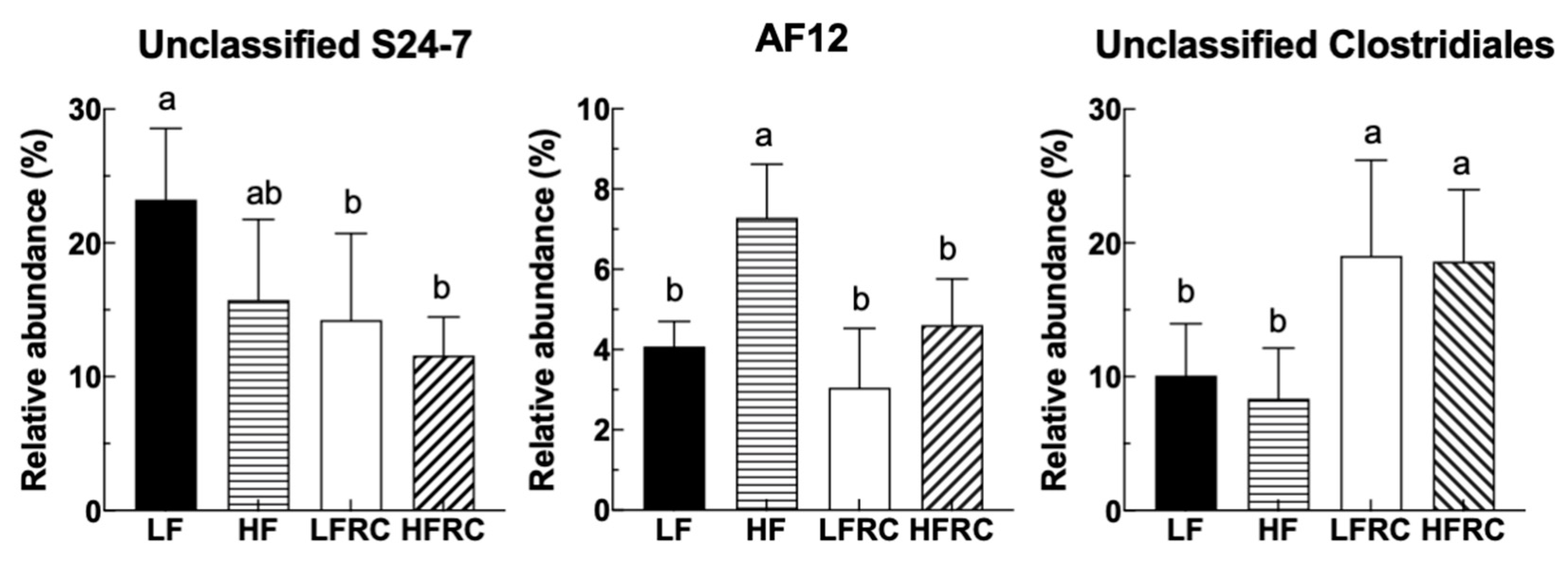
| Bacteroidia; Bacteroidales; S24-7 | Other | 23.24 ± 5.32 a | 15.74 ± 6.01 ab | 14.23 ± 6.49 b | 11.59 ± 2.87 b |
| Bacteroidia; Bacteroidales; Rikenellaceae | AF12 | 4.08 ± 0.62 b | 7.29 ± 1.33 a | 3.05 ± 1.48 b | 4.61 ± 1.15 b |
| Bacteroidia; Bacteroidales; [Odoribacteraceae] | Odoribacter | 7.60 ± 2.26 ab | 6.52 ± 1.88 b | 9.73 ± 2.49 a | 6.22 ± 1.60 b |
| Firmicutes | |||||
| Clostridia; Clostridiales; Ruminococcaceae | Oscillospira | 10.17 ± 1.97 b | 14.01 ± 2.74 ab | 9.45 ± 2.99 b | 16.80 ± 4.34 a |
| Clostridia; Clostridiales; Ruminococcaceae | Other | 5.17 ± 1.52 b | 8.70 ± 1.80 a | 4.19 ± 1.57 b | 7.61 ± 1.66 a |
| Clostridia; Clostridiales; Other | Other | 10.10 ± 3.87 b | 8.36 ± 3.78 b | 19.04 ± 7.16 a | 18.62 ± 5.36 a |
| Clostridia; Clostridiales; Lachnospiraceae | Coprococcus | 0.24 ± 0.05 a | 0.34 ± 0.26 a | 0.55 ± 0.22 a | 0.53 ± 0.22 b |
| Proteobacteria | |||||
| Deltaproteobacteria; Desulfovibrionales; Desulfovibrionaceae | Bilophila | 2.52 ± 0.60 a | 4.03 ± 0.47 a | 1.81 ± 0.66 b | 3.39 ± 0.64 b |
| Diet Group | Physiological Traits | Bacterial Taxa (Rank) | r a | p-Value b |
|---|---|---|---|---|
| LF | Fecal bile acid | Bacteroidaceae (Family) | 0.741 | 0.004 |
| Fecal bile acid | Bacteroides (Genus) | 0.741 | 0.002 | |
| HF | Hepatic free cholesterol | Bacteroidaceae (Family) | 0.684 | 0.003 |
| Hepatic free cholesterol | Bacteroides (Genus) | 0.684 | 0.001 | |
| LFRC | Hepatic. free. cholesterol | Oscillospira (Genus) | 0.316 | 0.046 |
| LDL | [Ruminococcus] (Genus) | 0.790 | 0.017 | |
| LDL | Gnavus (Species) | 0.790 | 0.017 | |
| HFRC | Hepatic triglycerides | Lachnospiraceae (Family) | 0.269 | 0.003 |
| Hepatic triglycerides | S24-7 (Family) | 0.243 | 0.036 | |
| Hepatic triglycerides | Coprococcus (Genus) | 0.718 | 0.017 | |
| Hepatic triglycerides | Oscillospira (Genus) | 0.134 | 0.048 |
| Diet Group | Module | Physiological Traits | r a | p-Value b |
|---|---|---|---|---|
| LF | 9 | Body weight | 0.84 | 0.02 |
| 7 | Hepatic cholesterol ester | 0.88 | 0.008 | |
| 7 | Hepatic free cholesterol | −0.92 | 0.003 | |
| 1 | Fecal bile acid | 0.81 | 0.03 | |
| HF | 10 | Body weight | −0.81 | 0.03 |
| 2 | VLDL | 0.83 | 0.02 | |
| 9 | VLDL | 0.89 | 0.007 | |
| 8 | Hepatic cholesterol ester | −0.79 | 0.04 | |
| 6 | Hepatic free cholesterol | 0.84 | 0.02 | |
| 7 | Fecal bile acid | 0.79 | 0.03 | |
| LFRC | 7 | Hepatic free cholesterol | −0.72 | 0.05 |
| HFRC | 6 | HDL | −0.74 | 0.04 |
| 8 | Hepatic triglycerides | −0.80 | 0.02 | |
| 5 | Hepatic free cholesterol | −0.73 | 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Xin, M.; Pham, Q.; Gao, Y.; Huang, H.; Jiang, X.; Li, R.W.; Yu, L.; Luo, Y.; Wang, J.; et al. Red Cabbage Modulates Composition and Co-Occurrence Networks of Gut Microbiota in a Rodent Diet-Induced Obesity Model. Foods 2024, 13, 85. https://doi.org/10.3390/foods13010085
Wu Y, Xin M, Pham Q, Gao Y, Huang H, Jiang X, Li RW, Yu L, Luo Y, Wang J, et al. Red Cabbage Modulates Composition and Co-Occurrence Networks of Gut Microbiota in a Rodent Diet-Induced Obesity Model. Foods. 2024; 13(1):85. https://doi.org/10.3390/foods13010085
Chicago/Turabian StyleWu, Yanbei, Mengmeng Xin, Quynhchi Pham, Yu Gao, Haiqiu Huang, Xiaojing Jiang, Robert W. Li, Liangli Yu, Yaguang Luo, Jing Wang, and et al. 2024. "Red Cabbage Modulates Composition and Co-Occurrence Networks of Gut Microbiota in a Rodent Diet-Induced Obesity Model" Foods 13, no. 1: 85. https://doi.org/10.3390/foods13010085