Microscale 3D Liver Bioreactor for In Vitro Hepatotoxicity Testing under Perfusion Conditions

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
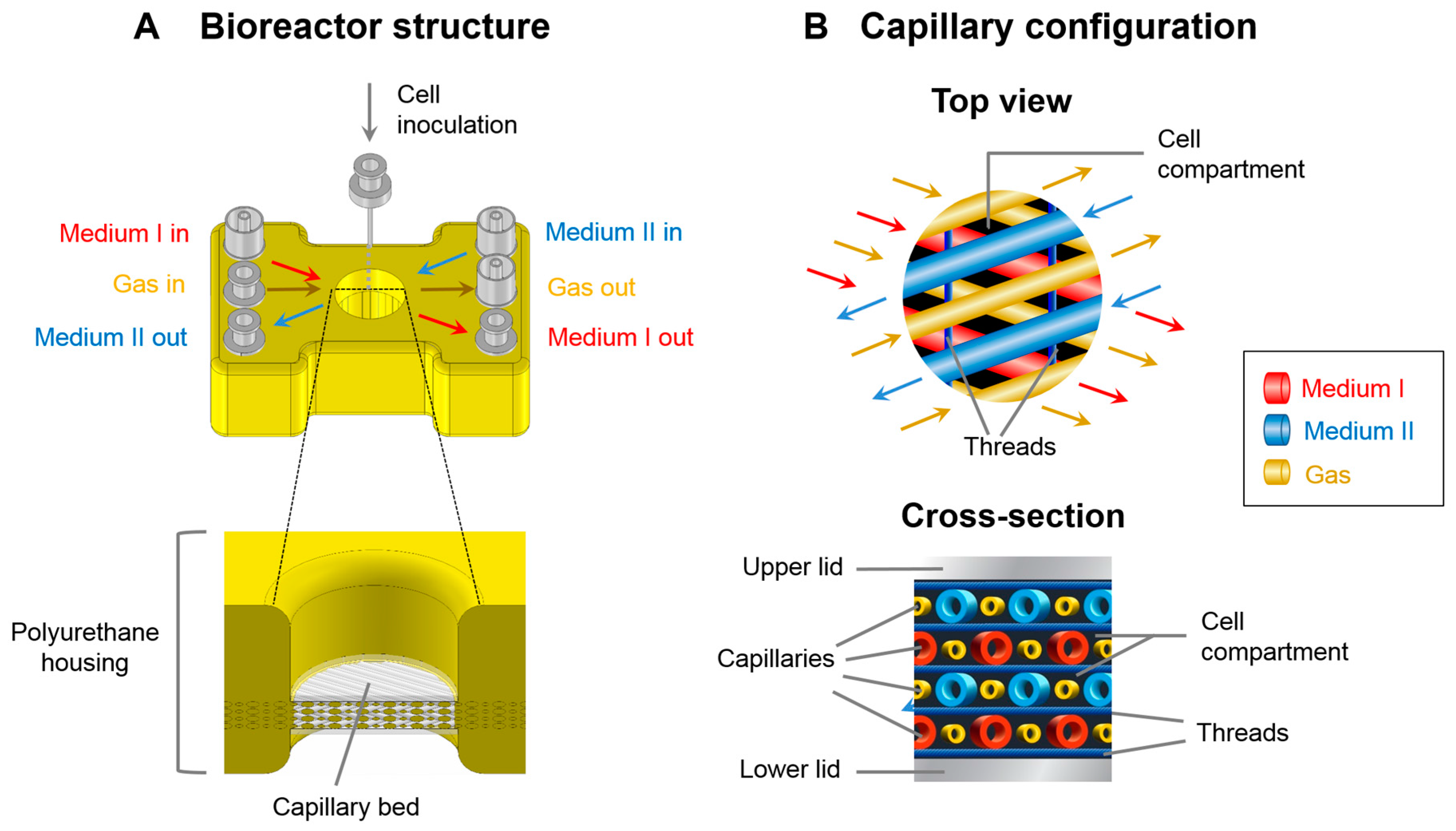
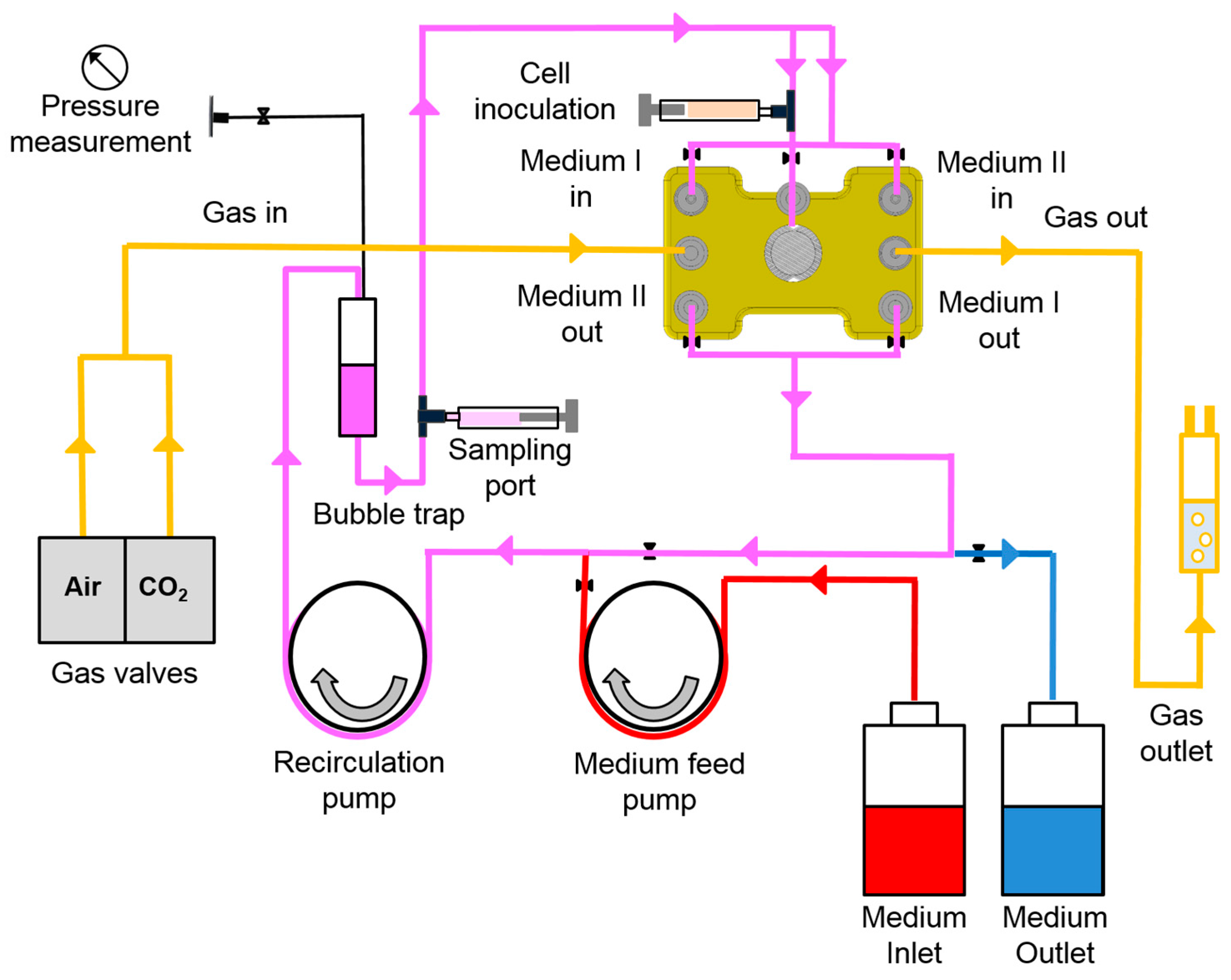
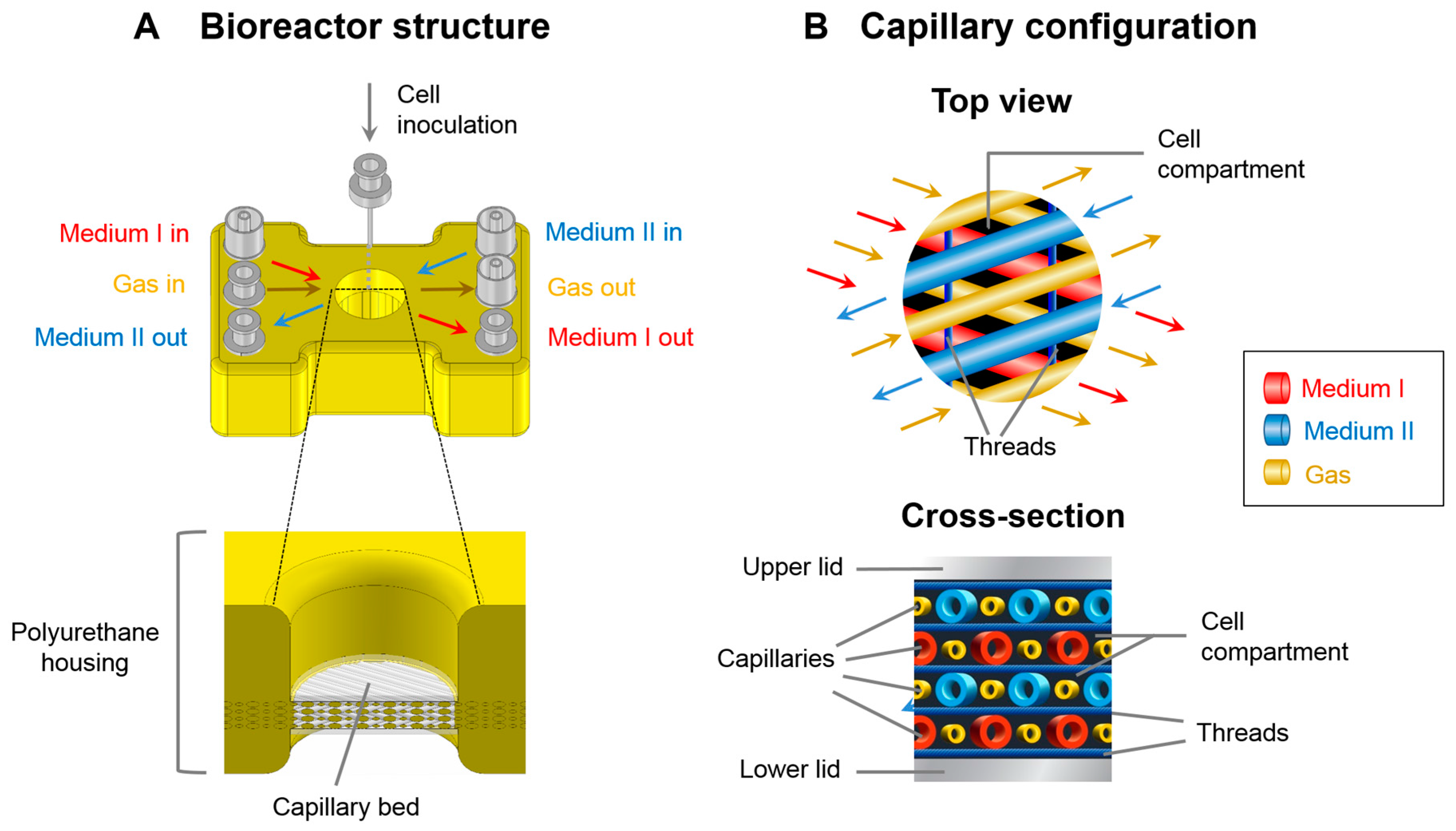
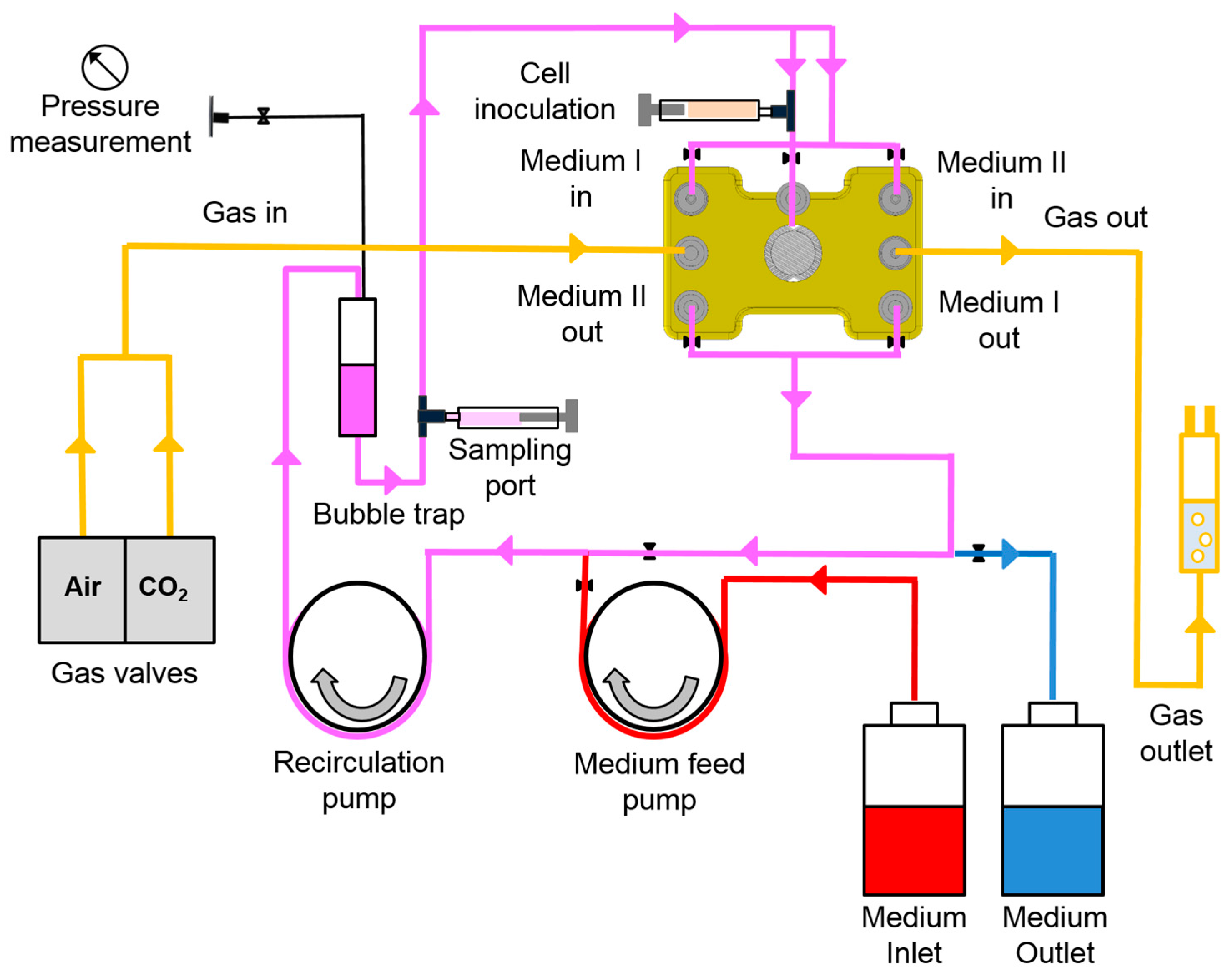
2.1. Bioreactor System
2.2. Primary Human Liver Cell Isolation
2.3. Bioreactor Operation
2.4. Clinical Chemistry Parameters
2.5. Acetaminophen (APAP) Application
2.6. Histological and Immunofluorescence Analysis
2.7. qRT-PCR
2.8. Statistics
3. Results
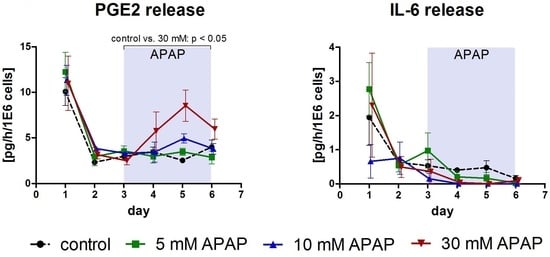
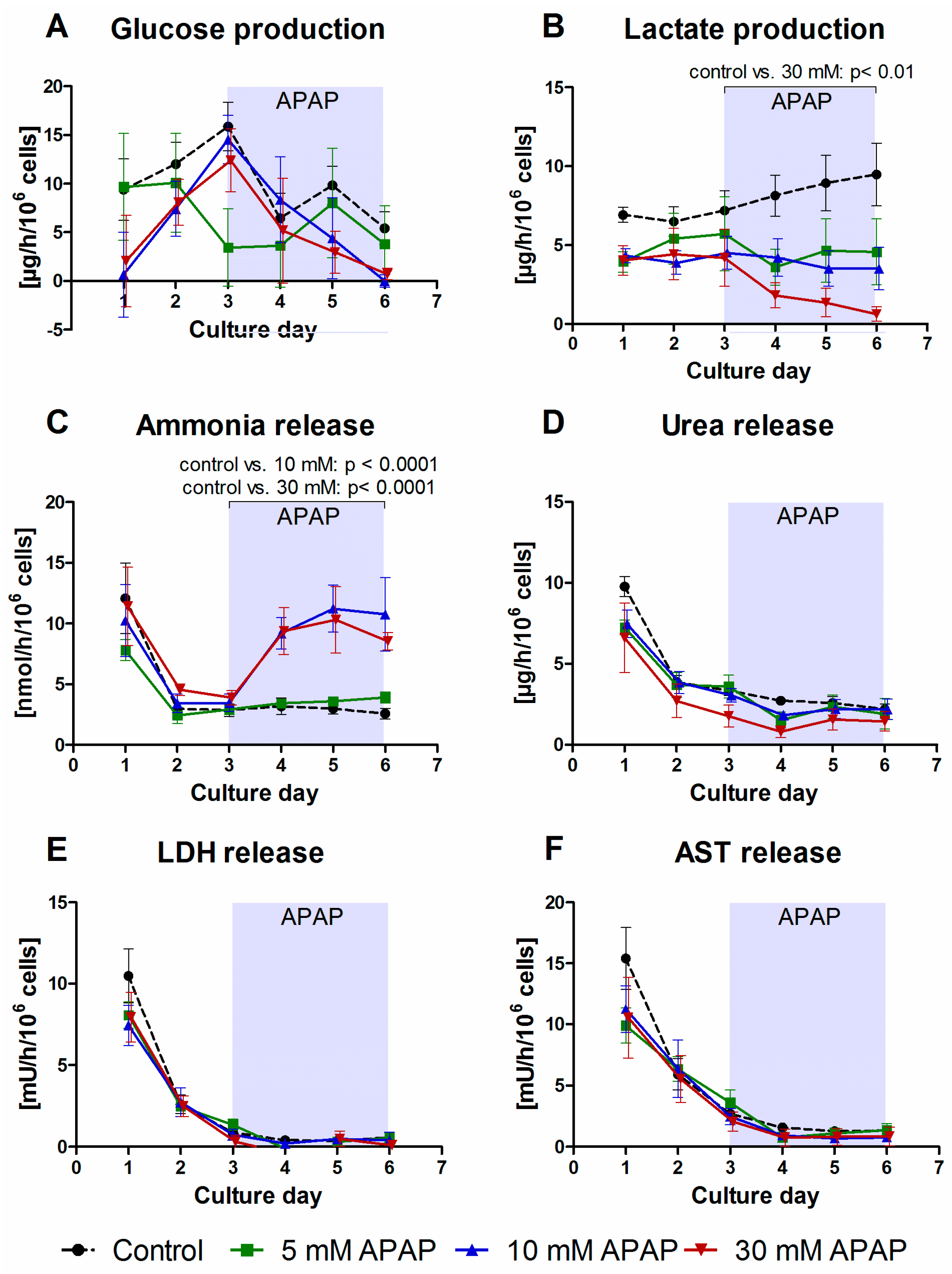
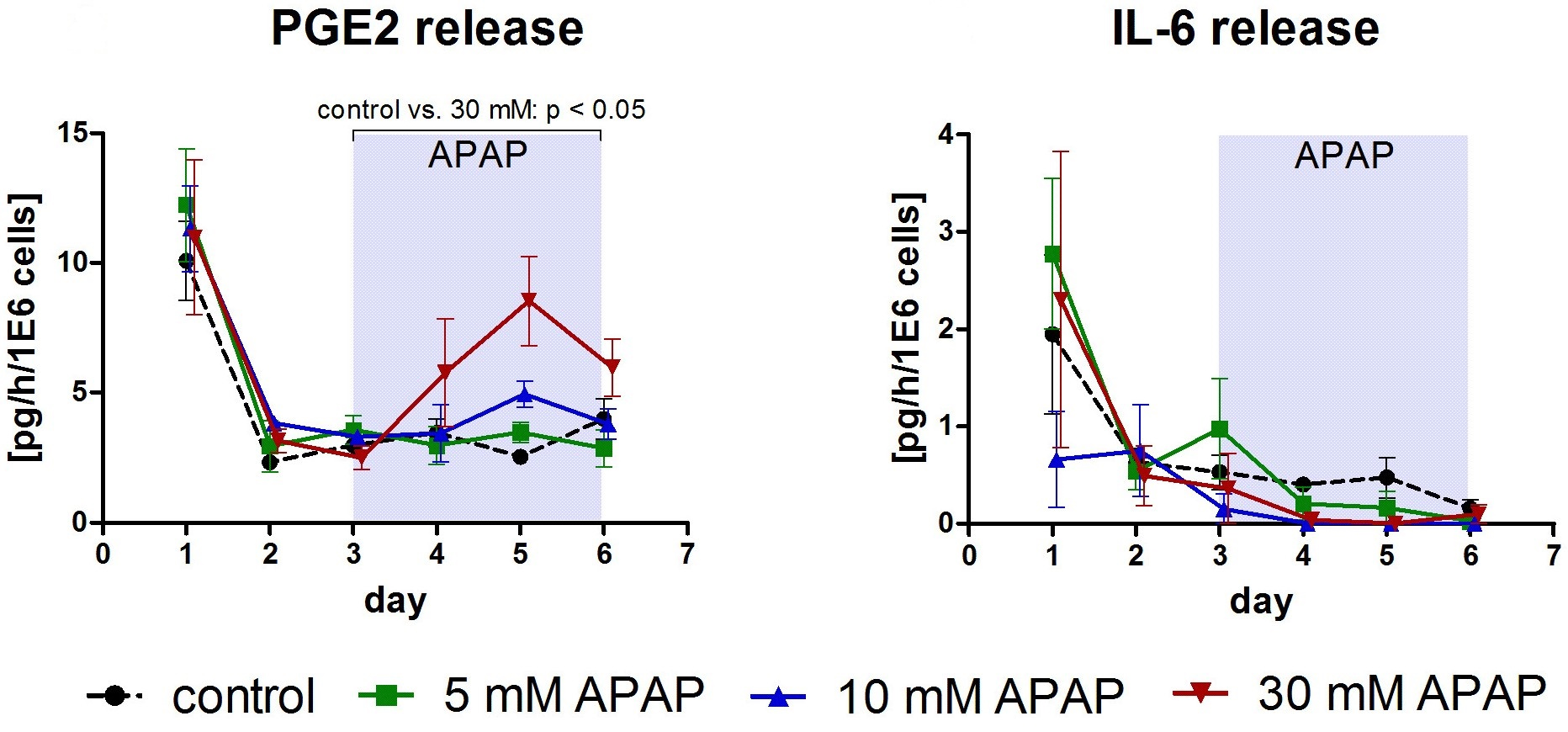
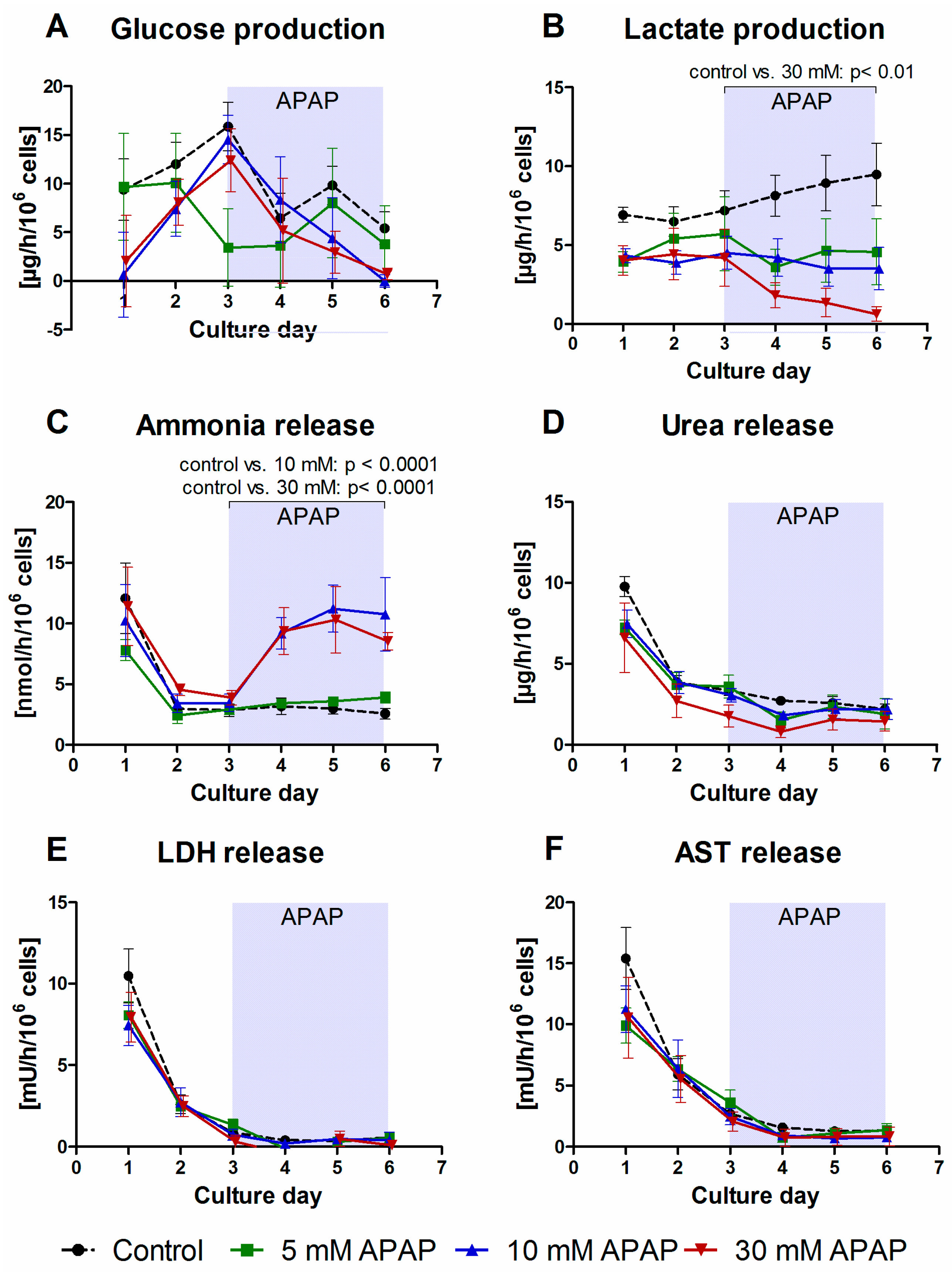
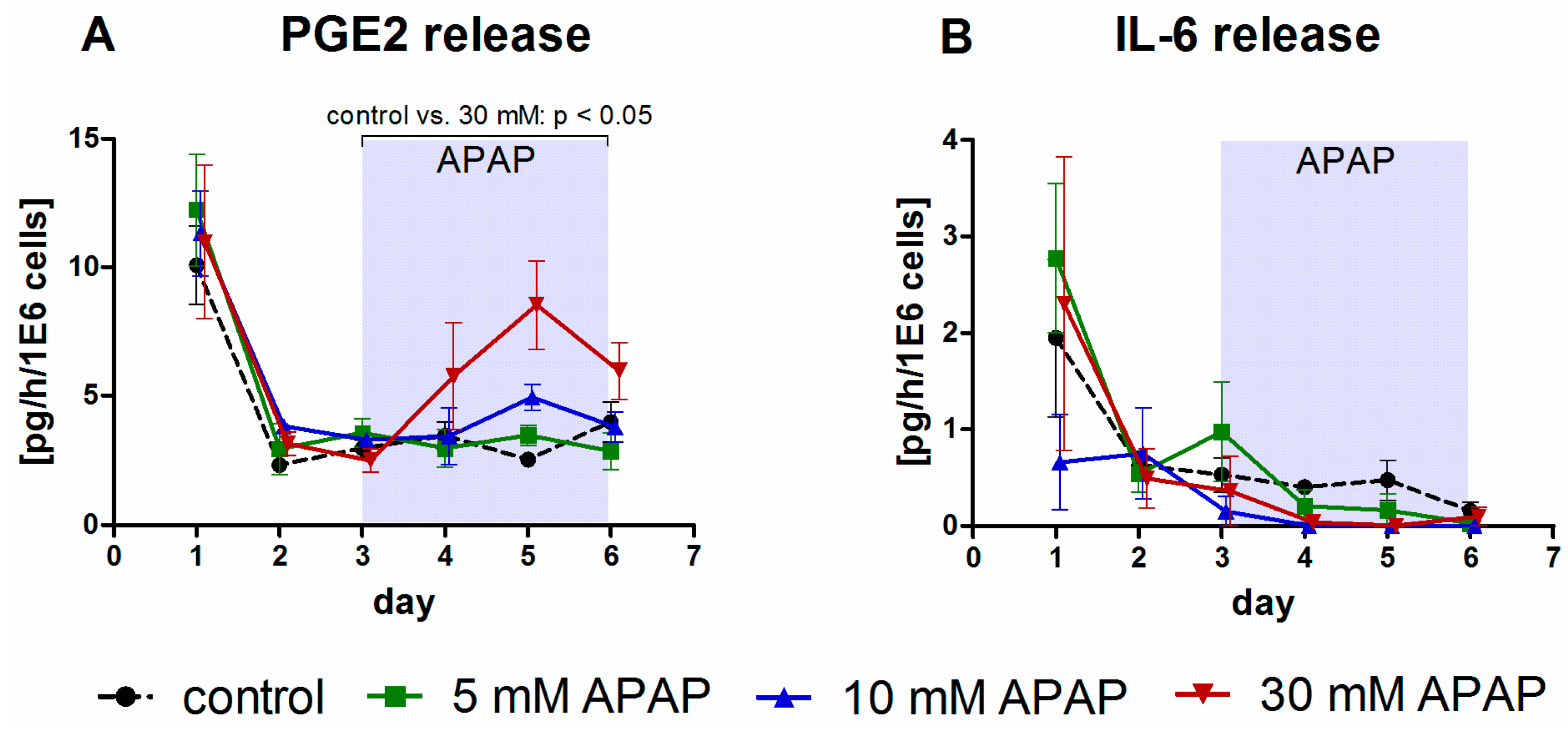
3.1. Clinical Chemistry Parameters
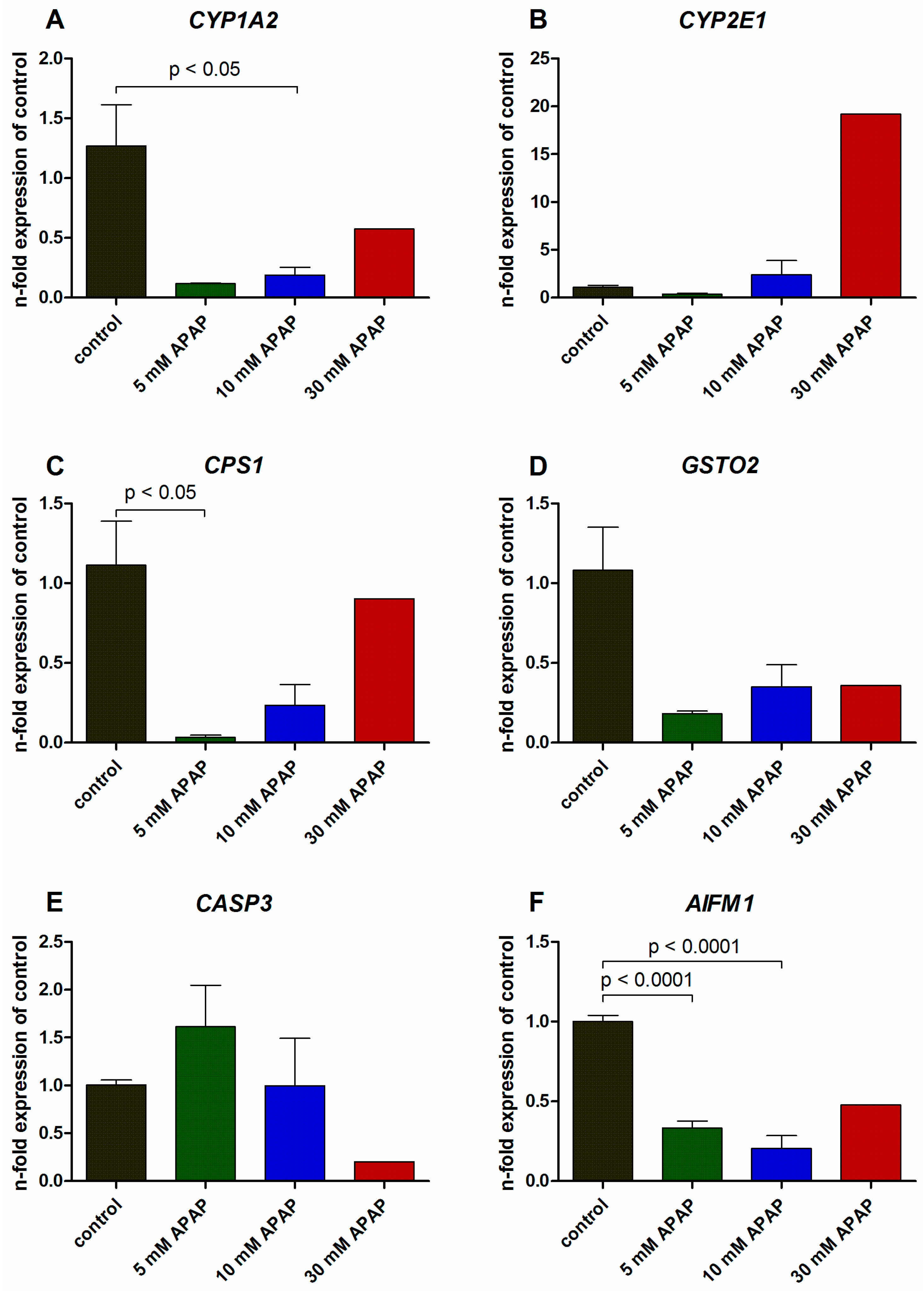
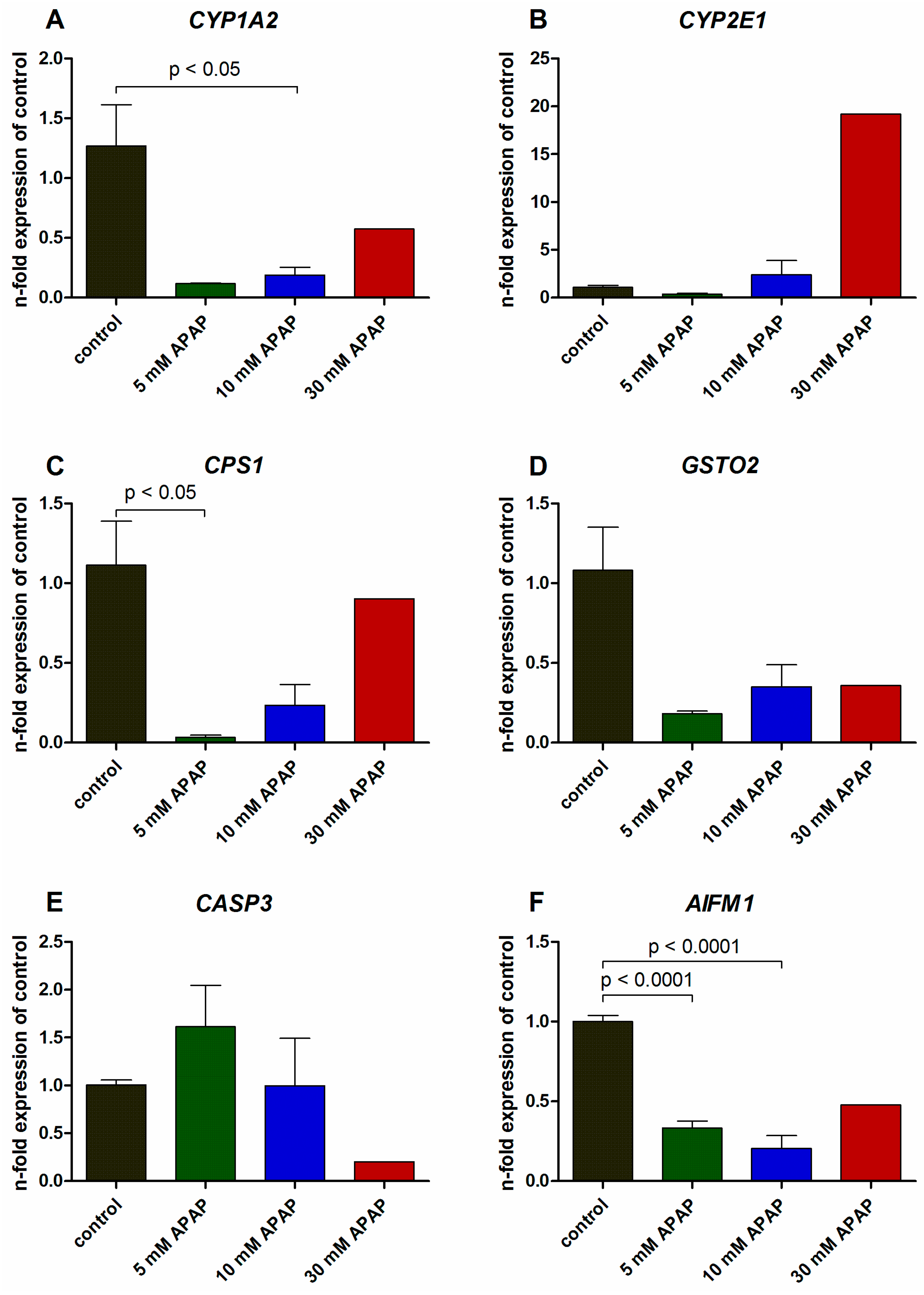
3.2. Gene Expression Analysis
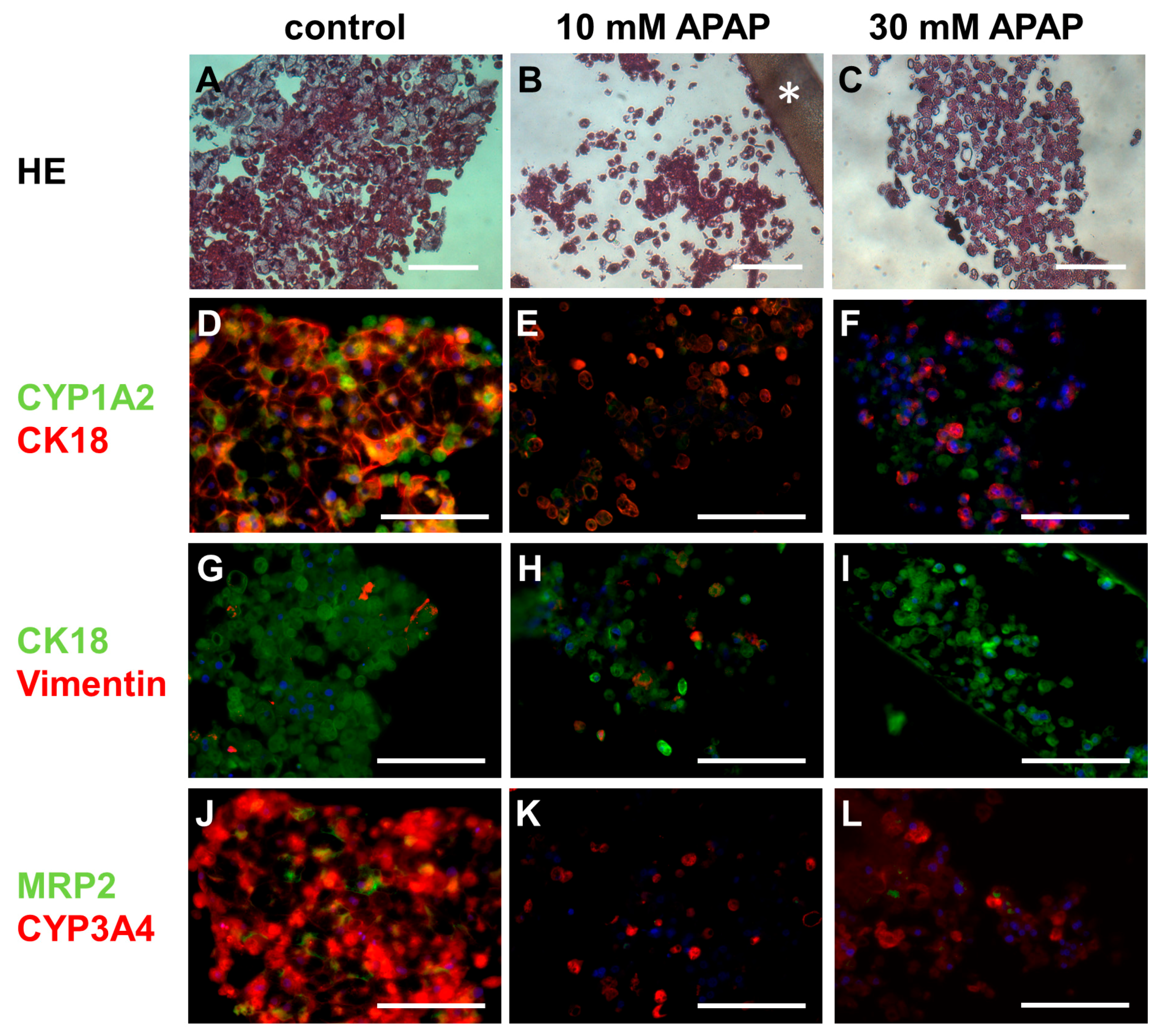
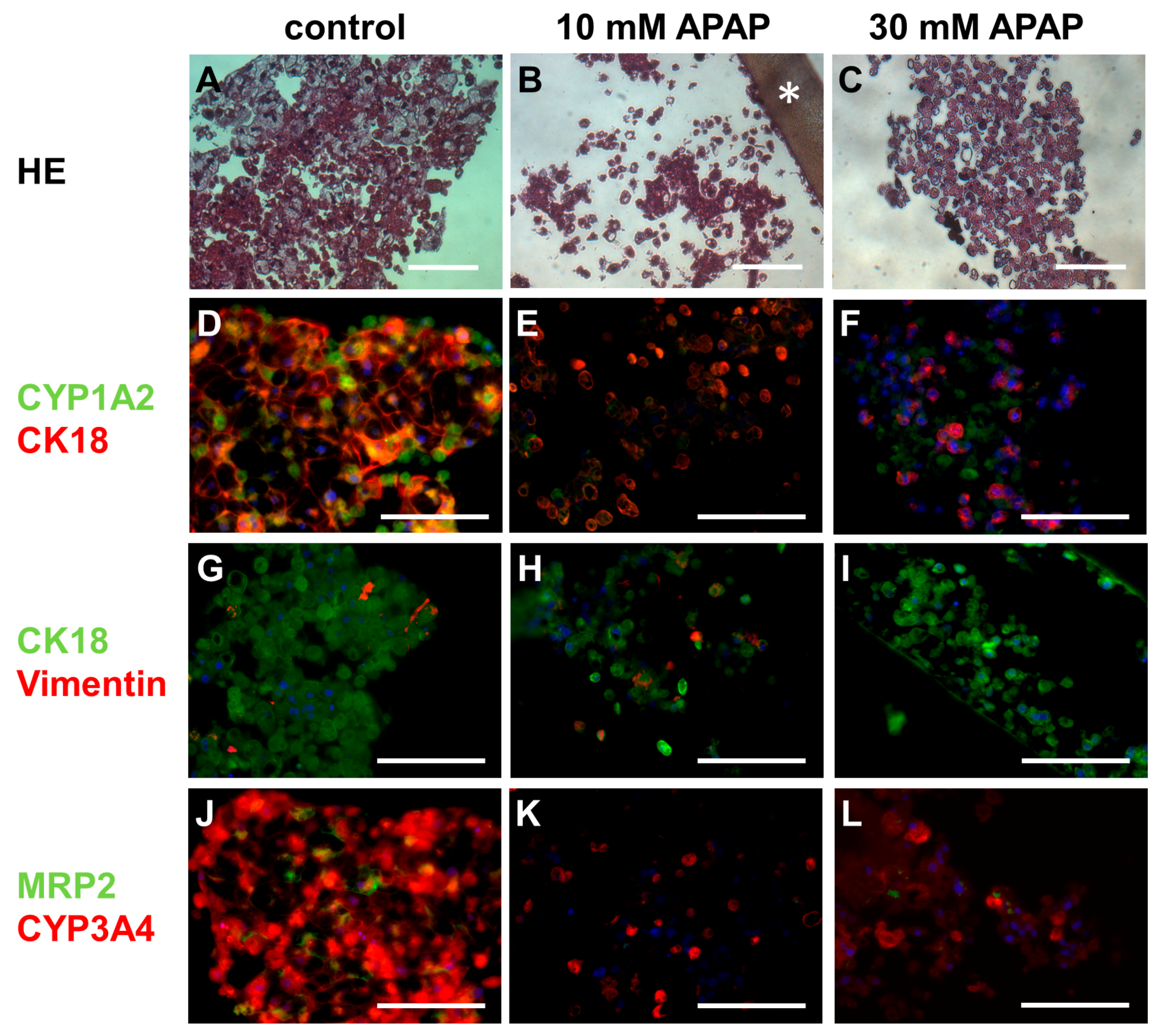
3.3. Histological and Immunohistochemical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhakuni, G.S.; Bedi, O.; Bariwal, J.; Deshmukh, R.; Kumar, P. Animal models of hepatotoxicity. Inflamm. Res. 2016, 65, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Vinken, M.; Jaeschke, H. Experimental models of hepatotoxicity related to acute liver failure. Toxicol. Appl. Pharmacol. 2016, 290, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; McNeill, J.H. To scale or not to scale: The principles of dose extrapolation. Br. J. Pharmacol. 2009, 157, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Elaut, G.; Henkens, T.; Papeleu, P.; Snykers, S.; Vinken, M.; Vanhaecke, T.; Rogiers, V. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Curr. Drug Metab. 2006, 7, 629–660. [Google Scholar] [CrossRef] [PubMed]
- Elliott, N.T.; Yuan, F. A review of three-dimensional in vitro tissue models for drug discovery and transport studies. J. Pharm. Sci. 2011, 100, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Schyschka, L.; Sánchez, J.J.; Wang, Z.; Burkhardt, B.; Müller-Vieira, U.; Zeilinger, K.; Bachmann, A.; Nadalin, S.; Damm, G.; Nussler, A.K. Hepatic 3D cultures but not 2D cultures preserve specific transporter activity for acetaminophen-induced hepatotoxicity. Arch. Toxicol. 2013, 87, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Hendriks, D.F.; Moro, S.M.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, T.; Ohta, K.; Nagao, T.; Kusumoto, K.; Koeda, A.; Ueda, T.; Jomura, T.; Ikeya, T.; Ozeki, E.; Wada, K.; et al. Evaluation of human hepatocytes cultured by three-dimensional spheroid systems for drug metabolism. Drug Metab. Pharmacokinet. 2014, 29, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.; Simmers, M.B.; Deering, T.G.; Berry, D.J.; Feaver, R.E.; Hastings, N.E.; Pruett, T.L.; LeCluyse, E.L.; Blackman, B.R.; Wamhoff, B.R. Hemodynamic flow improves rat hepatocyte morphology, function, and metabolic activity in vitro. Am. J. Physiol. Cell Physiol. 2013, 304, C1053–C1063. [Google Scholar] [CrossRef] [PubMed]
- De Bartolo, L.; Salerno, S.; Curcio, E.; Piscioneri, A.; Rende, M.; Morelli, S.; Tasselli, F.; Bader, A.; Drioli, E. Human hepatocyte functions in a crossed hollow fiber membrane bioreactor. Biomaterials 2009, 30, 2531–2543. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.U.R.; Geng, C.; Fu, M.; Yu, X.; Qin, K.; Liu, B. The role of microfluidics for organ on chip simulations. Bioengineering 2017, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Liu, J.R.; Patel, B.; Solomon, D.E.; Vaidya, B.; Gupta, V. Microfluidics-based 3D cell culture models: Utility in novel drug discovery and delivery research. Bioeng. Transl. Med. 2016, 1, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Zeilinger, K.; Schreiter, T.; Darnell, M.; Söderdahl, T.; Lübberstedt, M.; Dillner, B.; Knobeloch, D.; Nüssler, A.K.; Gerlach, J.C.; Andersson, T.B. Scaling down of a clinical three-dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies. Tissue Eng. Part C 2011, 17, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Knöspel, F.; Jacobs, F.; Freyer, N.; Damm, G.; De Bondt, A.; van den Wyngaert, I.; Snoeys, J.; Monshouwer, M.; Richter, M.; Strahl, N.; et al. In vitro model for hepatotoxicity studies based on primary human hepatocyte cultivation in a perfused 3D bioreactor system. Int. J. Mol. Sci. 2016, 17, 584. [Google Scholar] [CrossRef] [PubMed]
- Lübberstedt, M.; Müller-Vieira, U.; Biemel, K.M.; Darnell, M.; Hoffmann, S.A.; Knöspel, F.; Wönne, E.C.; Knobeloch, D.; Nüssler, A.K.; Gerlach, J.C.; et al. Serum-free culture of primary human hepatocytes in a miniaturized hollow-fibre membrane bioreactor for pharmacological in vitro studies. J. Tissue Eng. Regen. Med. 2015, 9, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.A.; Müller-Vieira, U.; Biemel, K.; Knobeloch, D.; Heydel, S.; Lübberstedt, M.; Nüssler, A.K.; Andersson, T.B.; Gerlach, J.C.; Zeilinger, K. Analysis of drug metabolism activities in a miniaturized liver cell bioreactor for use in pharmacological studies. Biotechnol. Bioeng. 2012, 109, 3172–3181. [Google Scholar] [CrossRef] [PubMed]
- Kegel, V.; Deharde, D.; Pfeiffer, E.; Zeilinger, K.; Seehofer, D.; Damm, G. Protocol for isolation of primary human hepatocytes and corresponding major populations of non-parenchymal liver cells. J. Vis. Exp. 2016, 109, e53069. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, E.; Kegel, V.; Zeilinger, K.; Hengstler, J.G.; Nüssler, A.K.; Seehofer, D.; Damm, G. Featured Article: Isolation, characterization, and cultivation of human hepatocytes and non-parenchymal liver cells. Exp. Biol. Med. 2015, 240, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Freyer, N.; Knöspel, F.; Strahl, N.; Amini, L.; Schrade, P.; Bachmann, S.; Damm, G.; Seehofer, D.; Jacobs, F.; Monshouwer, M.; et al. Hepatic differentiation of human induced pluripotent stem cells in a perfused three-dimensional multicompartment bioreactor. BioRes. Open Access 2016, 5, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. [Google Scholar] [CrossRef]
- Treyer, A.; Müsch, A. Hepatocyte polarity. Compr. Physiol. 2013, 3, 243–287. [Google Scholar] [CrossRef] [PubMed]
- Tormos, A.M.; Taléns-Visconti, R.; Bonora-Centelles, A.; Pérez, S.; Sastre, J. Oxidative stress triggers cytokinesis failure in hepatocytes upon isolation. Free Radic. Res. 2015, 49, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; McGill, M.R.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Forster, J.; Jaeschke, H. Mechanisms of acetaminophen-induced cell death in primary human hepatocytes. Toxicol. Appl. Pharmacol. 2014, 279, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Prill, S.; Jaeger, M.S.; Duschl, C. Long-term microfluidic glucose and lactate monitoring in hepatic cell culture. Biomicrofluidics 2014, 8, 034102. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.S.; Wills, L.P.; Schnellmann, R.G. Measurement of cell death in mammalian cells. Curr. Protoc. Pharmacol. 2012. [Google Scholar] [CrossRef]
- Silva, M.T. Secondary necrosis: The natural outcome of the complete apoptotic program. FEBS Lett. 2010, 584, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Jialal, I.; Sokoll, L.J. Clinical utility of lactate dehydrogenase: A historical perspective. Am. J. Clin. Pathol. 2015, 143, 158–159. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, W.M.; Reynolds, T.B. Serum lactic dehydrogenase in the differential diagnosis of acute hepatocellular injury. J. Clin. Gastroenterol. 1994, 19, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.L. Biochemical diagnosis of liver disease. Indian J. Clin. Biochem. 1999, 14, 59–90. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Pfund, W.P.; Andersen, M.E.; Thomas, R.S.; Clewell, H.J.; LeCluyse, E.L. Development of 3D dynamic flow model of human liver and its application to prediction of metabolic clearance of 7-ethoxycoumarin. Tissue Eng. Part C Methods 2014, 20, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Stolz, D.B.; Ellis, E.C.; Strom, S.C.; Michalopoulos, G.K.; Hengstler, J.G.; Runge, D. Long term cultures of primary human hepatocytes as an alternative to drugtesting in animals. ALTEX 2009, 26, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Bolleyn, J.; Rogiers, V.; Vanhaecke, T. Functionality testing of primary hepatocytes in culture by measuring urea synthesis. Methods Mol. Biol. 2015, 1250, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed]
- Norris, C.A.; He, M.; Kang, L.I.; Ding, M.Q.; Radder, J.E.; Haynes, M.M.; Yang, Y.; Paranjpe, S.; Bowen, W.C.; Orr, A.; et al. Synthesis of IL-6 by hepatocytes is a normal response to common hepatic stimuli. PLoS ONE 2014, 9, e96053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Manyike, P.T.; Kharasch, E.D.; Kalhorn, T.F.; Slattery, J.T. Contribution of CYP2E1 and CYP3A to acetaminophen reactive metabolite formation. Clin. Pharmacol. Ther. 2000, 67, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Buters, J.T.; Pineau, T.; Fernandez-Salguero, P.; Gonzalez, F.J. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J. Biol. Chem. 1996, 271, 12063–12067. [Google Scholar] [CrossRef] [PubMed]
- Patten, C.J.; Thomas, P.E.; Guy, R.L.; Lee, M.; Gonzalez, F.J.; Guengerich, F.P.; Yang, C.S. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem. Res. Toxicol. 1993, 6, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Raucy, J.L.; Lasker, J.M.; Lieber, C.S.; Black, M. Acetaminophen activation by human liver cytochromes P450IIE1 and P450IA2. Arch. Biochem. Biophys. 1989, 271, 270–283. [Google Scholar] [CrossRef]
- Laine, J.E.; Auriola, S.; Pasanen, M.; Juvonen, R.O. Acetaminophen bioactivation by human cytochrome P450 enzymes and animal microsomes. Xenobiotica 2009, 39, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sarich, T.; Kalhorn, T.; Magee, S.; Al-Sayegh, F.; Adams, S.; Slattery, J.; Goldstein, J.; Nelson, S.; Wright, J. The effect of omeprazole pretreatment on acetaminophen metabolism in rapid and slow metabolizers of S-mephenytoin. Clin. Pharmacol. Ther. 1997, 62, 21–28. [Google Scholar] [CrossRef]
- Khosravi, M.; Saadat, I.; Karimi, M.H.; Geramizadeh, B.; Saadat, M. Glutathione S-transferase omega 2 genetic polymorphism and risk of hepatic failure that lead to liver transplantation in iranian population. Int. J. Organ Transplant. Med. 2013, 4, 16–20. [Google Scholar] [PubMed]
- Chen, C.; Hennig, G.E.; Manautou, J.E. Hepatobiliary excretion of acetaminophen glutathione conjugate and its derivatives in transport-deficient (TR-) hyperbilirubinemic rats. Drug Metab. Dispos. 2003, 31, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Zhang, C.; Fu, L.; Zhang, D.G.; Wang, B.W.; Zhang, Z.H.; Chen, Y.H.; Lu, Y.; Chen, X.; Xu, D.X. Protective effect of rosiglitazone against acetaminophen-induced acute liver injury is associated with down-regulation of hepatic NADPH oxidases. Toxicol. Lett. 2017, 265, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Au, S.H.; Chamberlain, M.D.; Mahesh, S.; Sefton, M.V.; Wheeler, A.R. Hepatic organoids for microfluidic drug screening. Lab Chip 2014, 14, 3290–3299. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Grunseich, C.; Sevrioukova, I.F.; Schindler, A.; Horkayne-Szakaly, I.; Lamperti, C.; Landouré, G.; Kennerson, M.L.; Burnett, B.G.; Bönnemann, C.; et al. Cowchock syndrome is associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet. 2012, 91, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Malhi, H.; Mott, J.L.; Gores, G.J. Apoptosis and necrosis in the liver. Compr. Physiol. 2013, 3, 977–1010. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Bajt, M.L. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol. Sci. 2006, 89, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Hoffmaster, K.A.; Tian, X.; Zhao, R.; Polli, J.W.; Humphreys, J.E.; Webster, L.O.; Bridges, A.S.; Kalvass, J.C.; Brouwer, K.L. Multiple mechanisms are involved in the biliary excretion of acetaminophen sulfate in the rat: Role of MRP2 and BCRP1. Drug Metab. Dispos. 2005, 33, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhise, N.S.; Manoharan, V.; Massa, S.; Tamayol, A.; Ghaderi, M.; Miscuglio, M.; Lang, Q.; Shrike Zhang, Y.; Shin, S.R.; Calzone, G.; et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 2016, 8, 014101. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhao, L.; Zhou, E.M.; Xu, J.; Shen, S.; Wang, J. On-chip construction of liver lobule-like microtissue and its application for adverse drug reaction assay. Anal. Chem. 2016, 88, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Cairney, D.G.; Beckwith, H.K.; Al-Hourani, K.; Eddleston, M.; Bateman, D.N.; Dear, J.W. Plasma paracetamol concentration at hospital presentation has a dose-dependent relationship with liver injury despite prompt treatment with intravenous acetylcysteine. Clin. Toxicol. 2016, 54, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.C.; Block, G.D. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994, 272, 1845–1850. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Williams, C.D.; Ramachandran, A.; Bajt, M.L. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. 2012, 32, 8–20. [Google Scholar] [CrossRef] [PubMed]
- McCuskey, R.S. Sinusoidal endothelial cells as an early target for hepatic toxicants. Clin. Hemorheol. Microcirc. 2006, 34, 5–10. [Google Scholar] [PubMed]
- Kegel, V.; Pfeiffer, E.; Burkhardt, B.; Liu, J.L.; Zeilinger, K.; Nüssler, A.K.; Seehofer, D.; Damm, G. Subtoxic concentrations of hepatotoxic drugs lead to Kupffer cell activation in a human in vitro liver model: An approach to study DILI. Mediators Inflamm. 2015, 2015, 640631. [Google Scholar] [CrossRef] [PubMed]
- Prodanov, L.; Jindal, R.; Bale, S.S.; Hegde, M.; McCarty, W.J.; Golberg, I.; Bhushan, A.; Yarmush, M.L.; Usta, O.B. Long-term maintenance of a microfluidic 3D human liver sinusoid. Biotechnol. Bioeng. 2016, 113, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Rennert, K.; Steinborn, S.; Gröger, M.; Ungerböck, B.; Jank, A.M.; Ehgartner, J.; Nietzsche, S.; Dinger, J.; Kiehntopf, M.; Funke, H.; et al. A microfluidically perfused three dimensional human liver model. Biomaterials 2015, 71, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.W.; Khetani, S.R.; Bhatia, S.N. In vitro zonation and toxicity in a hepatocyte bioreactor. Toxicol. Sci. 2005, 84, 110–119. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Set Values during Operation |
|---|---|
| Recirculation rate | 1 mL/min |
| Feed rate | 0.6 mL/h (0–24 h) |
| 0.2 mL/h (from 24 h on) | |
| Gas flow rate | 4 mL/min |
| Concentration of CO2 in supplied gas mixture | 3–6% 1 |
| Temperature in bioreactor chamber | 38 °C |
| pH value | 7.35–7.45 |
| Parameter | Required Volume | Variation Coefficient |
|---|---|---|
| Glucose | 100 µL in total | 4% |
| Lactate | 3% | |
| LDH | 250 µL in total | 1% |
| AST | 4% | |
| ALT | 2.9% | |
| GLDH | 0.8% | |
| Urea | 1% | |
| Ammonia | 250 µL | 2.2% |
| PGE2 | 200 µL | n.a. |
| IL-6 | 200 µL | n.a. |
| Gene Symbol | Gene Name | Assay ID |
|---|---|---|
| AIFM1 | Apoptosis-inducing factor, mitochondria-associated, 1 | Hs00377585_m1 |
| CASP3 | Caspase 3, apoptosis-related cysteine peptidase | Hs00234387_m1 |
| CPS1 | Carbamoyl-phosphate synthase 1, mitochondrial | Hs00157048_m1 |
| CYP1A2 | Cytochrome P450, family 1, subfamily A, polypeptide 2 | Hs00167927_m1 |
| CYP2E1 | Cytochrome P450, family 2, subfamily E, polypeptide 1 | Hs00559368_m1 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | Hs03929097_g1 |
| GSTO2 | Glutathione S-transferase omega 2 | Hs01598184_m1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freyer, N.; Greuel, S.; Knöspel, F.; Gerstmann, F.; Storch, L.; Damm, G.; Seehofer, D.; Foster Harris, J.; Iyer, R.; Schubert, F.; et al. Microscale 3D Liver Bioreactor for In Vitro Hepatotoxicity Testing under Perfusion Conditions. Bioengineering 2018, 5, 24. https://doi.org/10.3390/bioengineering5010024
Freyer N, Greuel S, Knöspel F, Gerstmann F, Storch L, Damm G, Seehofer D, Foster Harris J, Iyer R, Schubert F, et al. Microscale 3D Liver Bioreactor for In Vitro Hepatotoxicity Testing under Perfusion Conditions. Bioengineering. 2018; 5(1):24. https://doi.org/10.3390/bioengineering5010024
Chicago/Turabian StyleFreyer, Nora, Selina Greuel, Fanny Knöspel, Florian Gerstmann, Lisa Storch, Georg Damm, Daniel Seehofer, Jennifer Foster Harris, Rashi Iyer, Frank Schubert, and et al. 2018. "Microscale 3D Liver Bioreactor for In Vitro Hepatotoxicity Testing under Perfusion Conditions" Bioengineering 5, no. 1: 24. https://doi.org/10.3390/bioengineering5010024