
Chitosan–Cellulose Multifunctional Hydrogel Beads: Design, Characterization and Evaluation of Cytocompatibility with Breast Adenocarcinoma and Osteoblast Cells


 ,
, 
Abstract
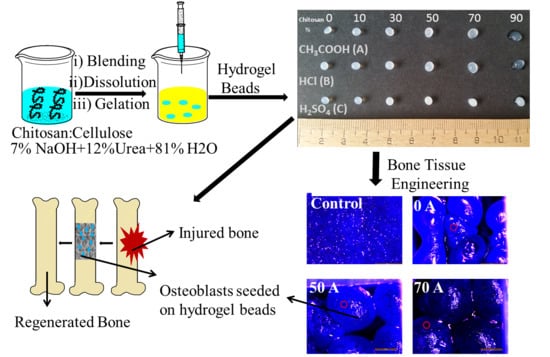
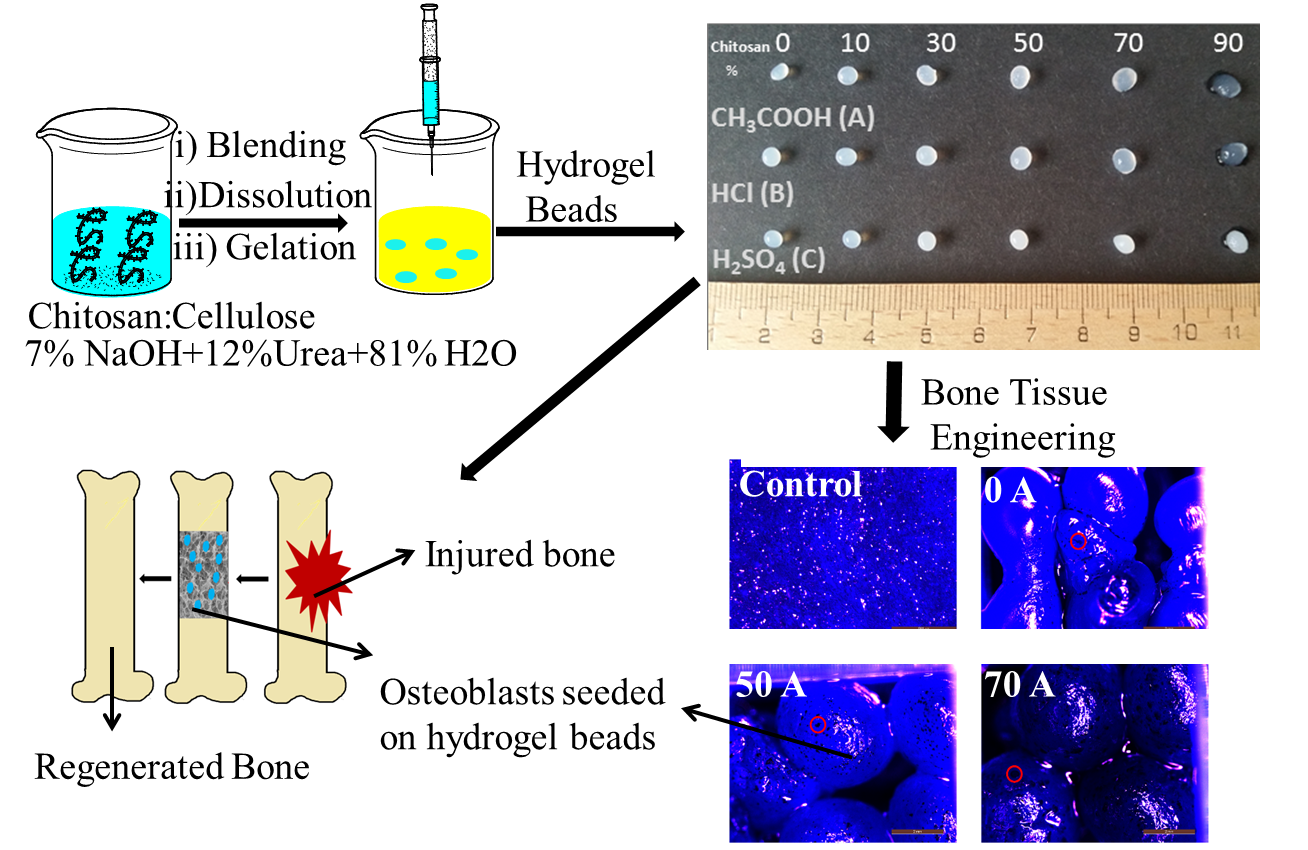{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Chitosan-Cellulose Hydrogel Beads
2.2.2. Ftir and Raman Spectroscopy
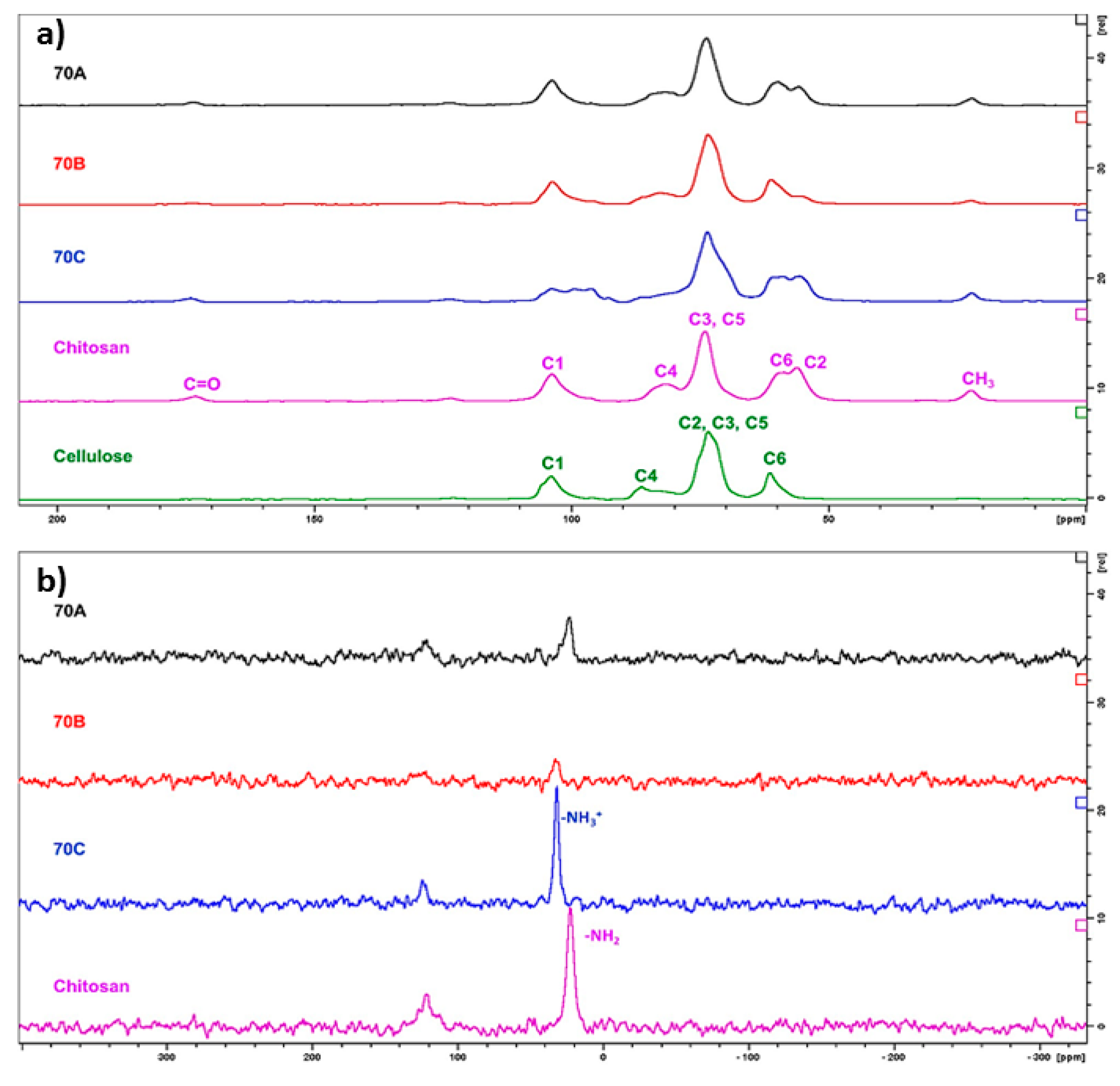
2.2.3. Solid State 13C and 15N NMR Spectroscopy
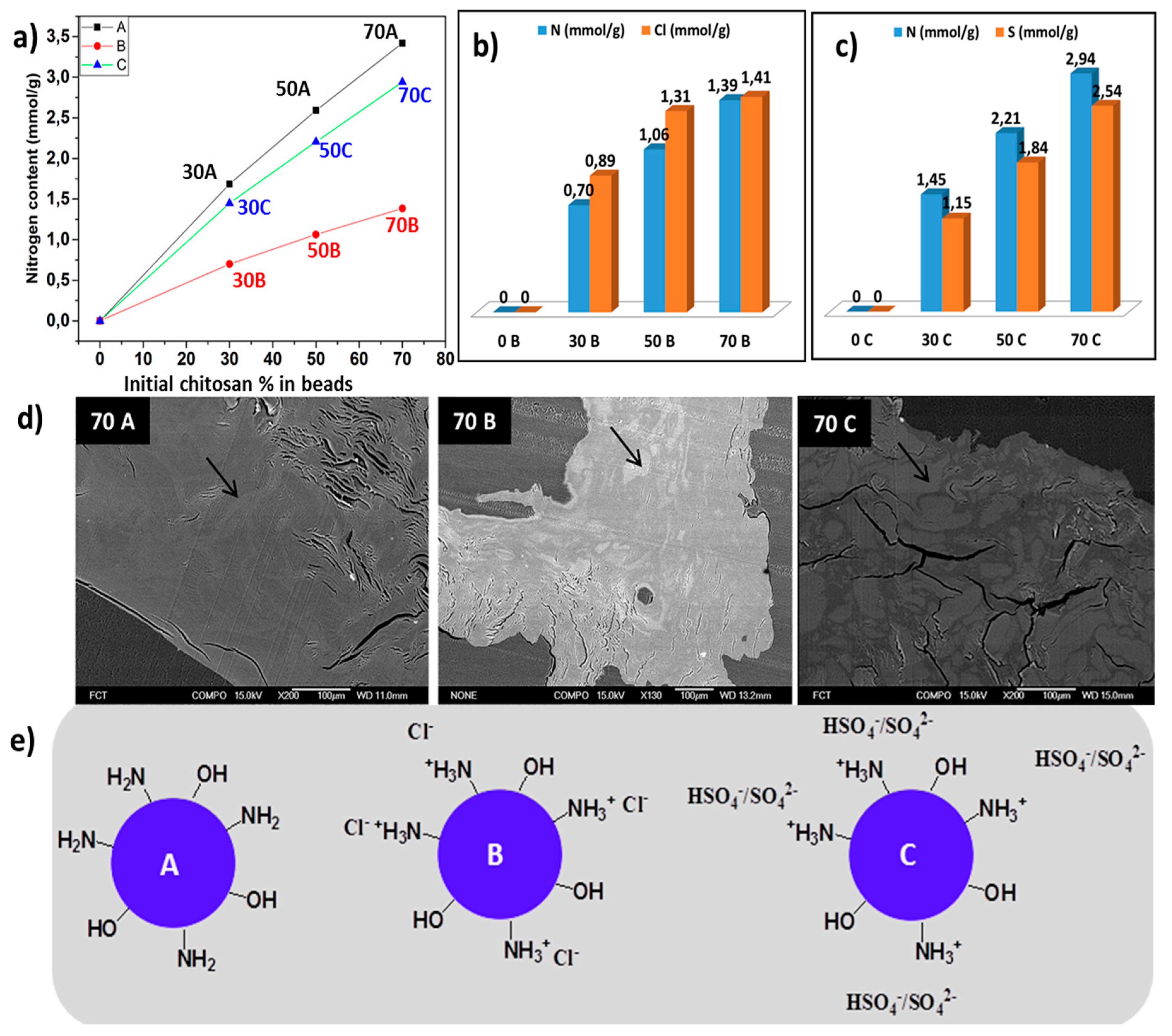
2.2.4. Elemental Analysis
2.2.5. XRD Analysis
2.2.6. Scanning Electron Microscopy-Energy Dispersive Spectroscopy
2.2.7. Cytocompatibility Evaluation of Chitosan-Cellulose Hydrogel Beads Coagulated in Different Acids with MDA-MB-231 (Human Breast Adenocarcinoma) Cells
2.2.8. Cytocompatibility Evaluation of Chitosan-Cellulose Hydrogel Beads Coagulated in 2 M Acetic Acid with Osteoblast Cells
2.2.9. Cell Attachment Testing of Chitosan-Cellulose Hydrogel Beads Coagulated in 2 M Acetic Acid with Osteoblast Cells
2.3.10. Statistical Analysis
3. Results and Discussions
3.1. Effect of Coagulating Medium on the Mechanism of the Chitosan-Cellulose Hydrogel Beads Formation
3.2. Attenuated Total Reflectance–Fourier Transform Infra-Red (ATR–FTIR) and Raman Spectroscopic Analysis
3.3. Solid-State 13C and 15N Nuclear Magnetic Resonance
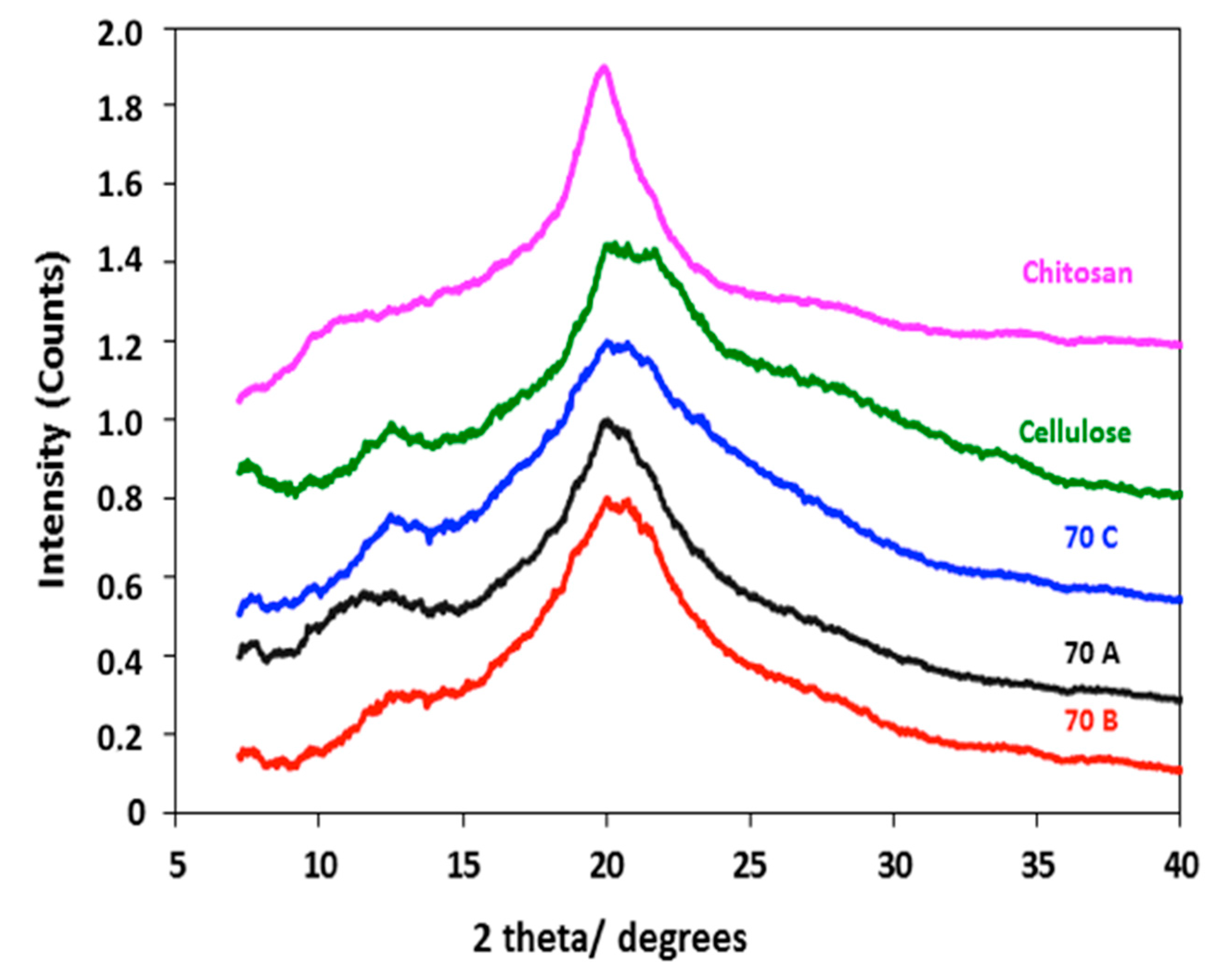
3.4. XRD Analysis
3.5. Scanning Electron Microscopic Analysis (SEM)
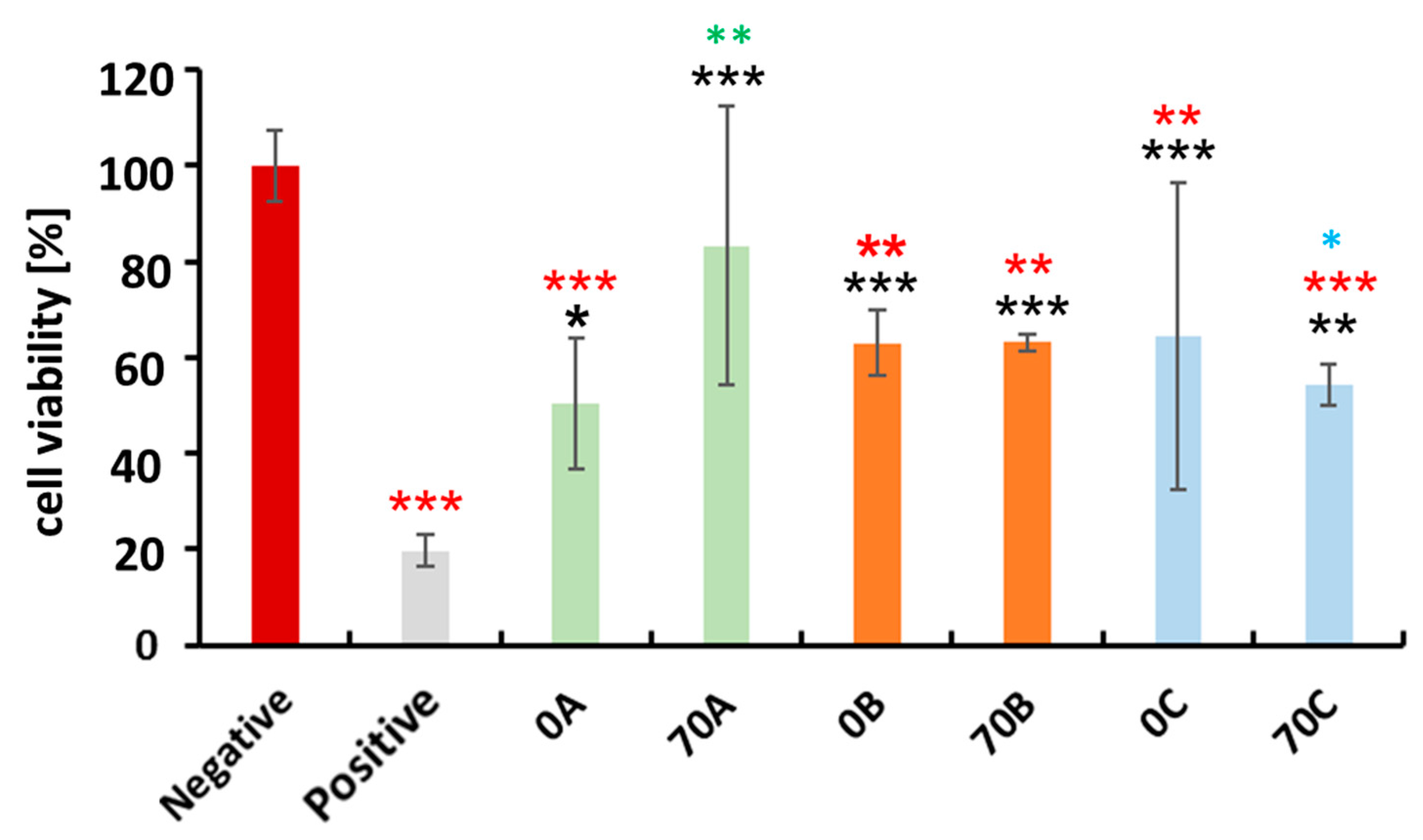
3.6. Cytocompatibility Evaluation of Chitosan-Cellulose Hydrogel Beads with MDA-MB-231 Cells (Human Breast Adenocarcinoma—A Soft Tissue Organ)
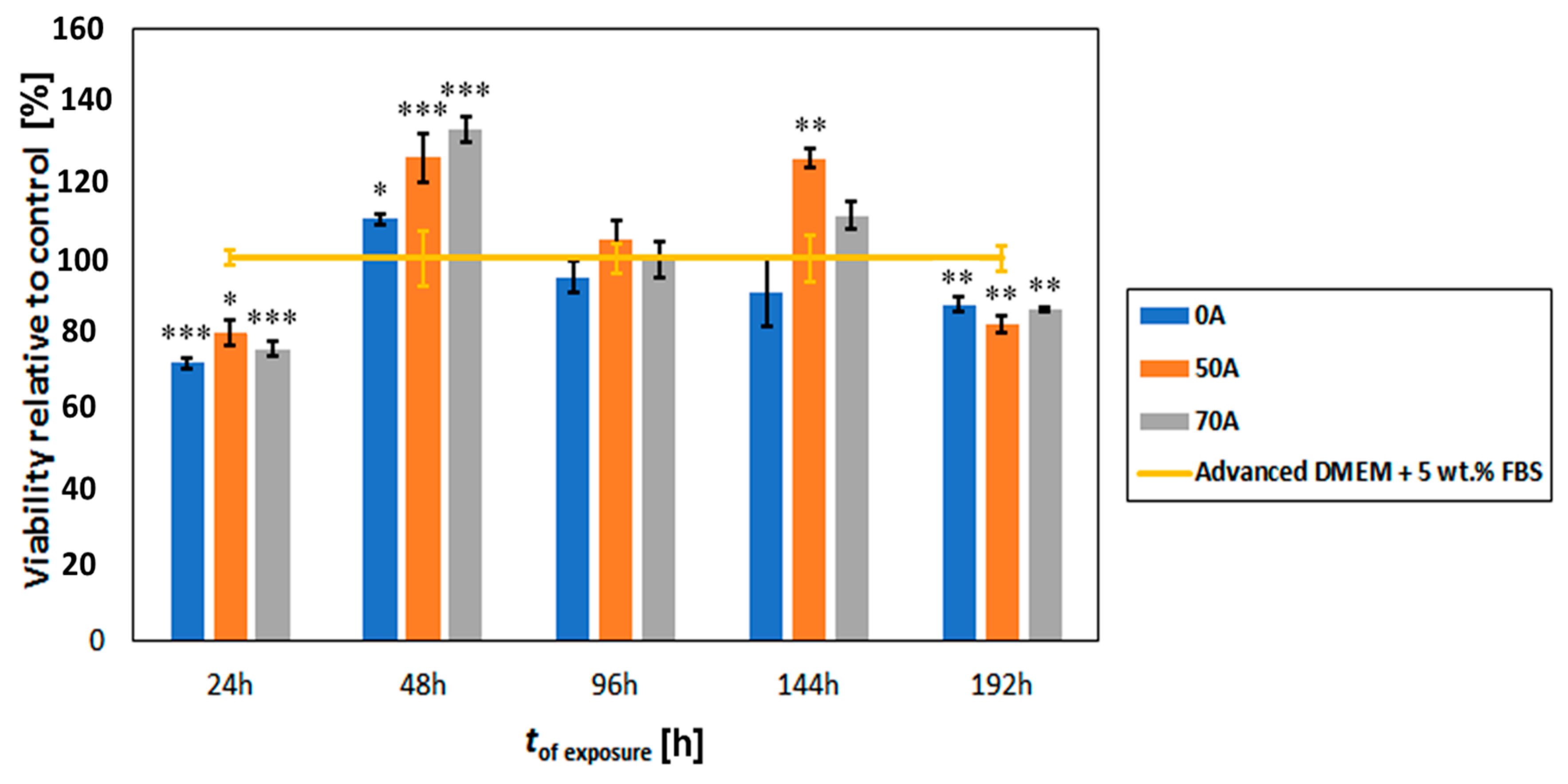
3.7. Cytocompatibility Evaluation of Chitosan-Cellulose Beads Coagulated in Acetic Acid with Osteoblast Cells (Hard Tissue)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mano, J.; Silva, G.; Azevedo, H.; Malafaya, P.; Sousa, R.; Silva, S.; Boesel, L.F.; Oliveira, J.M.; Santos, T.C.; Marques, A.P.; et al. Natural origin biodegradable systems in tissue engineering and regenerative medicine: Present status and some moving trends. J. R. Soc. Interface 2007, 4, 999–1030. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.; Ng, T.; Wong, J.; Chan, W. Chitosan: An update on potential biomedical and pharmaceutical applications. Mar. Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef] [PubMed]
- Malmström, E.; Carlmark, A. Controlled grafting of cellulose fibres—An outlook beyond paper and cardboard. Polym. Chem. 2012, 3, 1702–1713. [Google Scholar] [CrossRef]
- Vieira, M.; da Silva, M.; dos Santos, L.; Beppu, M. Natural-based plasticizers and biopolymer films: A review. Eur. Polym. J. 2011, 47, 254–263. [Google Scholar] [CrossRef]
- Gericke, M.; Trygg, J.; Fardim, P. Functional Cellulose Beads: Preparation, Characterization, and Applications. Chem. Rev. 2013, 113, 4812–4836. [Google Scholar] [CrossRef] [PubMed]
- Ravi Kumar, M. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Chaudhari, A.; Vig, K.; Baganizi, D.; Sahu, R.; Dixit, S.; Dennis, V.; Singh, S.R.; Pillai, S.R. Future prospects for scaffolding methods and biomaterials in skin tissue engineering: A Review. Int. J. Mol. Sci. 2016, 17, 1974. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yang, C.; Zhou, Z.; Chen, S.; Li, F. Characterization of biodegradable poly(lactic acid) porous scaffolds prepared using selective enzymatic degradation for tissue engineering. RSC Adv. 2017, 7, 34063–34070. [Google Scholar] [CrossRef]
- Pääkkö, M.; Vapaavuori, J.; Silvennoinen, R.; Kosonen, H.; Ankerfors, M.; Lindström, T.; Berglundc, L.A.; Ikkala, O. Long and entangled native cellulose I nanofibers allow flexible aerogels and hierarchically porous templates for functionalities. Soft Matter 2008, 4, 2492–2499. [Google Scholar] [CrossRef]
- Kim, I.; Seo, S.; Moon, H.; Yoo, M.; Park, I.; Kim, B.; Cho, C. Chitosan and its derivatives for tissue engineering applications. Biotechnol. Adv. 2008, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vázquez, M.; Vega-Ruiz, B.; Ramos-Zúñiga, R.; Saldaña-Koppel, D.; Quiñones-Olvera, L. Chitosan and its potential use as a scaffold for tissue engineering in regenerative medicine. BioMed Res. Int. 2015, 2015, 821279. [Google Scholar] [CrossRef] [PubMed]
- Finšgar, M.; Uzunalić, A.; Stergar, J.; Gradišnik, L.; Maver, U. Novel chitosan/diclofenac coatings on medical grade stainless steel for hip replacement applications. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Al-Munajjed, A.; Plunkett, N.; Gleeson, J.; Weber, T.; Jungreuthmayer, C.; Levingstone, T.; Hammer, J.; O’Brien, F.J. Development of a biomimetic collagen-hydroxyapatite scaffold for bone tissue engineering using a SBF immersion technique. J. Biomed. Mater. Res. Part B Appl. Biomater. 2009, 90B, 584–591. [Google Scholar] [CrossRef] [PubMed]
- VandeVord, P.; Matthew, H.; DeSilva, S.; Mayton, L.; Wu, B.; Wooley, P. Evaluation of the Cytocompatibility of a chitosan scaffold in mice. J. Biomed. Mater. Res. 2001, 59, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Costa-Pinto, A.; Reis, R.; Neves, N. Scaffolds based bone tissue engineering: The role of chitosan. Tissue Eng. Part B Rev. 2011, 17, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Ma, L. Collagen/chitosan porous scaffolds with improved biostability for skin tissue engineering. Biomaterials 2003, 24, 4833–4841. [Google Scholar] [CrossRef]
- Mwale, F.; Iordanova, M.; Demers, C.; Steffen, T.; Roughley, P.; Antoniou, J. Biological evaluation of chitosan salts cross-linked to genipin as a cell scaffold for disk tissue engineering. Tissue Eng. 2005, 11, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.; Tsai, S.; Ho, M.; Wang, D.; Liu, C.; Hsieh, C.; Tseng, H.; Hsieh, H. Analysis of freeze-gelation and cross-linking processes for preparing porous chitosan scaffolds. Carbohydr. Polym. 2007, 67, 124–132. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, Q.; Lin, D.; Yao, S. Effect and mechanism of sodium chloride on the formation of chitosan–cellulose sulfate–tripolyphosphate crosslinked beads. Soft Matter 2013, 9, 10354–10363. [Google Scholar] [CrossRef]
- Chan, B.; Leong, K. Scaffolding in tissue engineering: General approaches and tissue-specific considerations. Eur. Spine J. 2008, 17, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Sharma, S.; Liu, W.; Mu, W.; Liu, W.; Zhang, X.; Deng, Y. Aerogel Microspheres from Natural Cellulose Nanofibrils and Their Application as Cell Culture Scaffold. Biomacromolecules 2014, 15, 2540–2547. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, R.; Prabaharan, M.; Sudheesh Kumar, P.; Nair, S.; Tamura, H. Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol. Adv. 2011, 29, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Madihally, S.; Matthew, H. Porous chitosan scaffolds for tissue engineering. Biomaterials 1999, 20, 1133–1142. [Google Scholar] [CrossRef]
- Shen, X.; Shamshina, J.L.; Berton, P.; Gurau, G.; Rogers, R.D. Hydrogels based on cellulose and chitin: Fabrication, properties, and applications. Green Chem. 2016, 18, 53–75. [Google Scholar] [CrossRef]
- Li, N.; Bai, N. Copper adsorption on chitosan-cellulose hydrogel beads: Behaviors and mechanisms. Sep. Purif. Technol. 2005, 42, 237–247. [Google Scholar] [CrossRef]
- Twu, Y.K.; Huang, H.I.; Chang, S.Y.; Wang, S.L. Preparation and sorption activity of chitosan/cellulose blend beads. Carbohydr. Polym. 2003, 54, 425–430. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, H.; Liu, C.; Jiang, Y.; Yu, G.; Mu, X.; Wang, X. Magnetic cellulose–chitosan hydrogels prepared from ionic liquids as reusable adsorbent for removal of heavy metal ions. Chem. Commun. 2012, 48, 7350–7352. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhang, L.; Zhou, J.; Guo, S. Cellulose/chitin beads for adsorption of heavy metals in aqueous solution. Water Res. 2004, 38, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Trygg, J.; Fardim, P. Enhancement of cellulose dissolution in water-based solvent via ethanol–hydrochloric acid pretreatment. Cellulose 2011, 18, 987–994. [Google Scholar] [CrossRef]
- Van de Loosdrecht, A.A.; Beelen, R.H.J.; Ossenkoppele, G.J.; Broekhoven, M.G.; Langenhuijsen, M.M.A.C. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 1994, 174, 311–320. [Google Scholar] [CrossRef]
- Myllytie, P.; Salmi, J.; Laine, J. The influence of pH on the adsorption and interaction of chitosan with cellulose. Bioresources 2009, 4, 1647–1662. [Google Scholar]
- Cui, Z.; Xiang, Y.; Si, J.; Yang, M.; Zhang, Q.; Zhang, T. Ionic interactions between sulfuric acid and chitosan membranes. Carbohydr. Polym. 2008, 73, 111–116. [Google Scholar] [CrossRef]
- Larkin, P. Infrared and Raman Spectroscopy: Principles and Spectral Interpretation; Elsevier: Amsterdam, The Netherlands, 2011; pp. 110–130. [Google Scholar]
- Kono, H.; Yunoki, S.; Shikano, T.; Fujiwara, M.; Erata, T.; Takai, M. CP/MAS13C NMR Study of cellulose and cellulose derivatives. 1. Complete assignment of the CP/MAS13C NMR spectrum of the native cellulose. J. Am. Chem. Soc. 2002, 124, 7506–7511. [Google Scholar] [CrossRef] [PubMed]
- Heux, L.; Brugnerotto, J.; Desbrières, J.; Versali, M.; Rinaudo, M. Solid state NMR for determination of degree of acetylation of chitin and chitosan. Biomacromolecules 2000, 1, 746–751. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trivedi, P.; Saloranta-Simell, T.; Maver, U.; Gradišnik, L.; Prabhakar, N.; Smått, J.-H.; Mohan, T.; Gericke, M.; Heinze, T.; Fardim, P. Chitosan–Cellulose Multifunctional Hydrogel Beads: Design, Characterization and Evaluation of Cytocompatibility with Breast Adenocarcinoma and Osteoblast Cells. Bioengineering 2018, 5, 3. https://doi.org/10.3390/bioengineering5010003
Trivedi P, Saloranta-Simell T, Maver U, Gradišnik L, Prabhakar N, Smått J-H, Mohan T, Gericke M, Heinze T, Fardim P. Chitosan–Cellulose Multifunctional Hydrogel Beads: Design, Characterization and Evaluation of Cytocompatibility with Breast Adenocarcinoma and Osteoblast Cells. Bioengineering. 2018; 5(1):3. https://doi.org/10.3390/bioengineering5010003
Chicago/Turabian StyleTrivedi, Poonam, Tiina Saloranta-Simell, Uroš Maver, Lidija Gradišnik, Neeraj Prabhakar, Jan-Henrik Smått, Tamilselvan Mohan, Martin Gericke, Thomas Heinze, and Pedro Fardim. 2018. "Chitosan–Cellulose Multifunctional Hydrogel Beads: Design, Characterization and Evaluation of Cytocompatibility with Breast Adenocarcinoma and Osteoblast Cells" Bioengineering 5, no. 1: 3. https://doi.org/10.3390/bioengineering5010003
APA StyleTrivedi, P., Saloranta-Simell, T., Maver, U., Gradišnik, L., Prabhakar, N., Smått, J.-H., Mohan, T., Gericke, M., Heinze, T., & Fardim, P. (2018). Chitosan–Cellulose Multifunctional Hydrogel Beads: Design, Characterization and Evaluation of Cytocompatibility with Breast Adenocarcinoma and Osteoblast Cells. Bioengineering, 5(1), 3. https://doi.org/10.3390/bioengineering5010003