
Canine Mammary Carcinomas: A Comparative Analysis of Altered Gene Expression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Canine Mammary Tumors (CMTs)
3. Cell Cycle Regulators: A Classic Repertoire of Tumor Suppressors
3.1. CDK Inhibitors Form a Repertoire of Tumor Suppressor Proteins

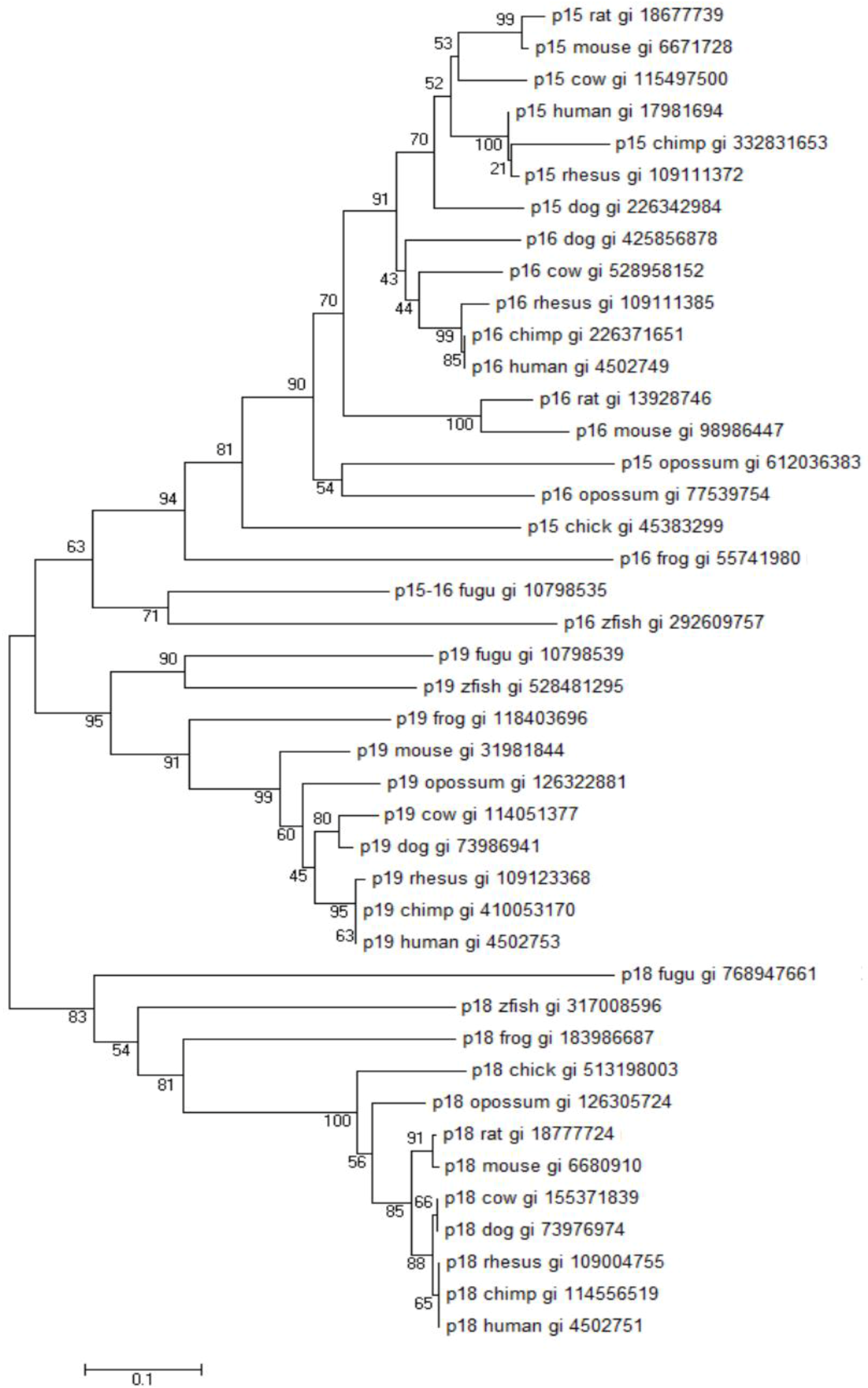
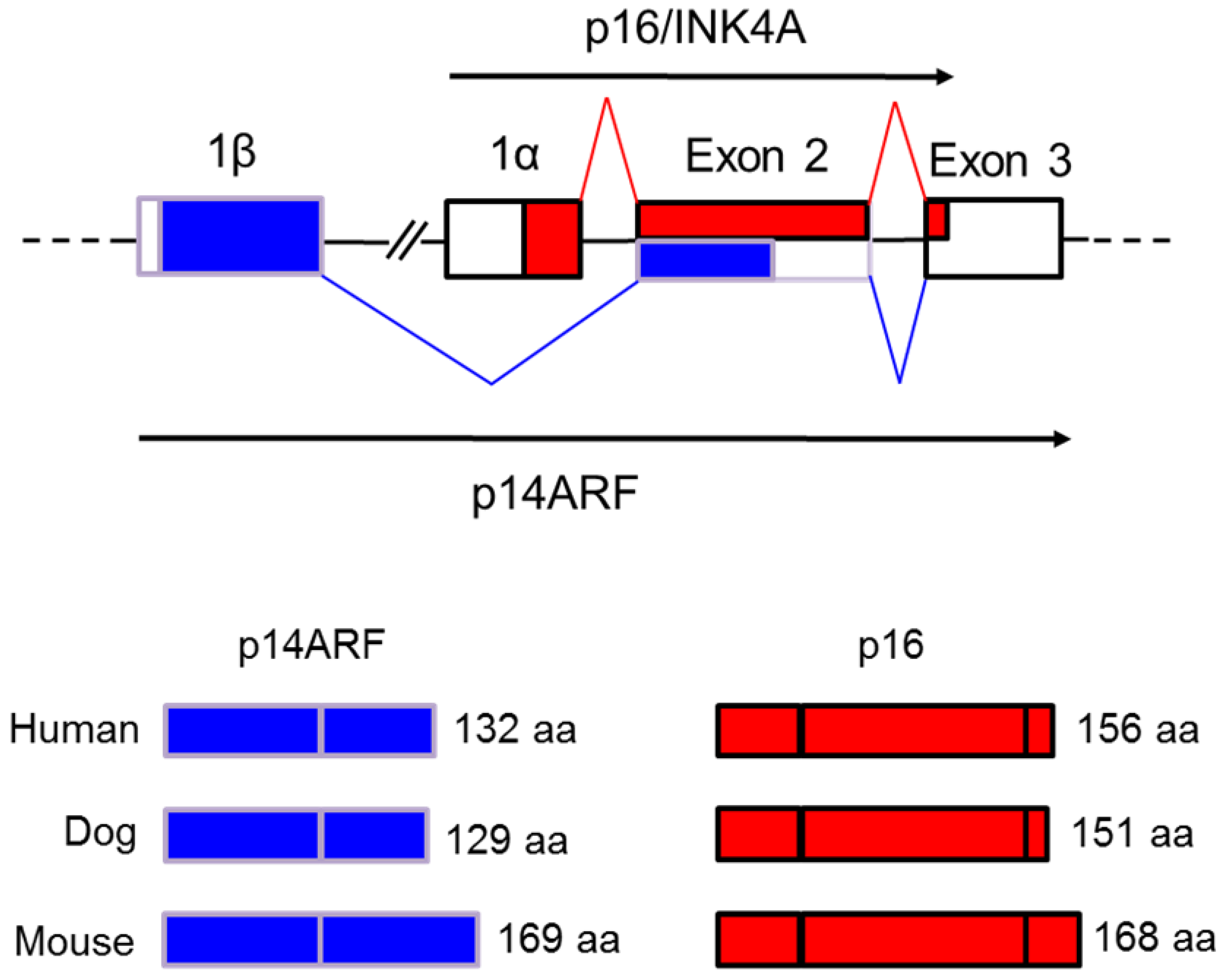
3.2. Evolutionary History, Genomic Localization and Structure of the INK4A/ARF Locus

3.3. Roles of INK4A/ARF Encoded Regulators in the Cell Cycle and Cancer

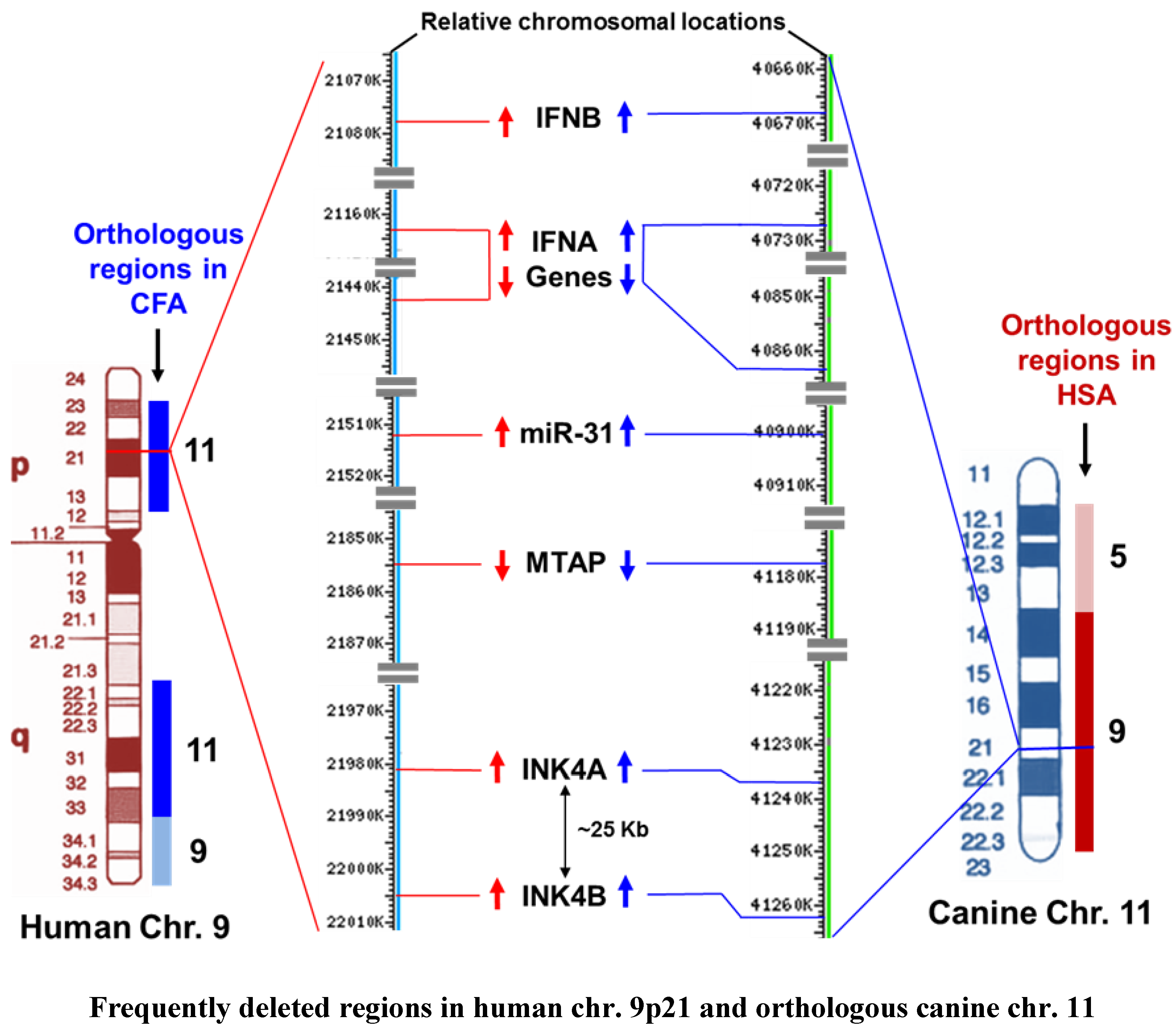
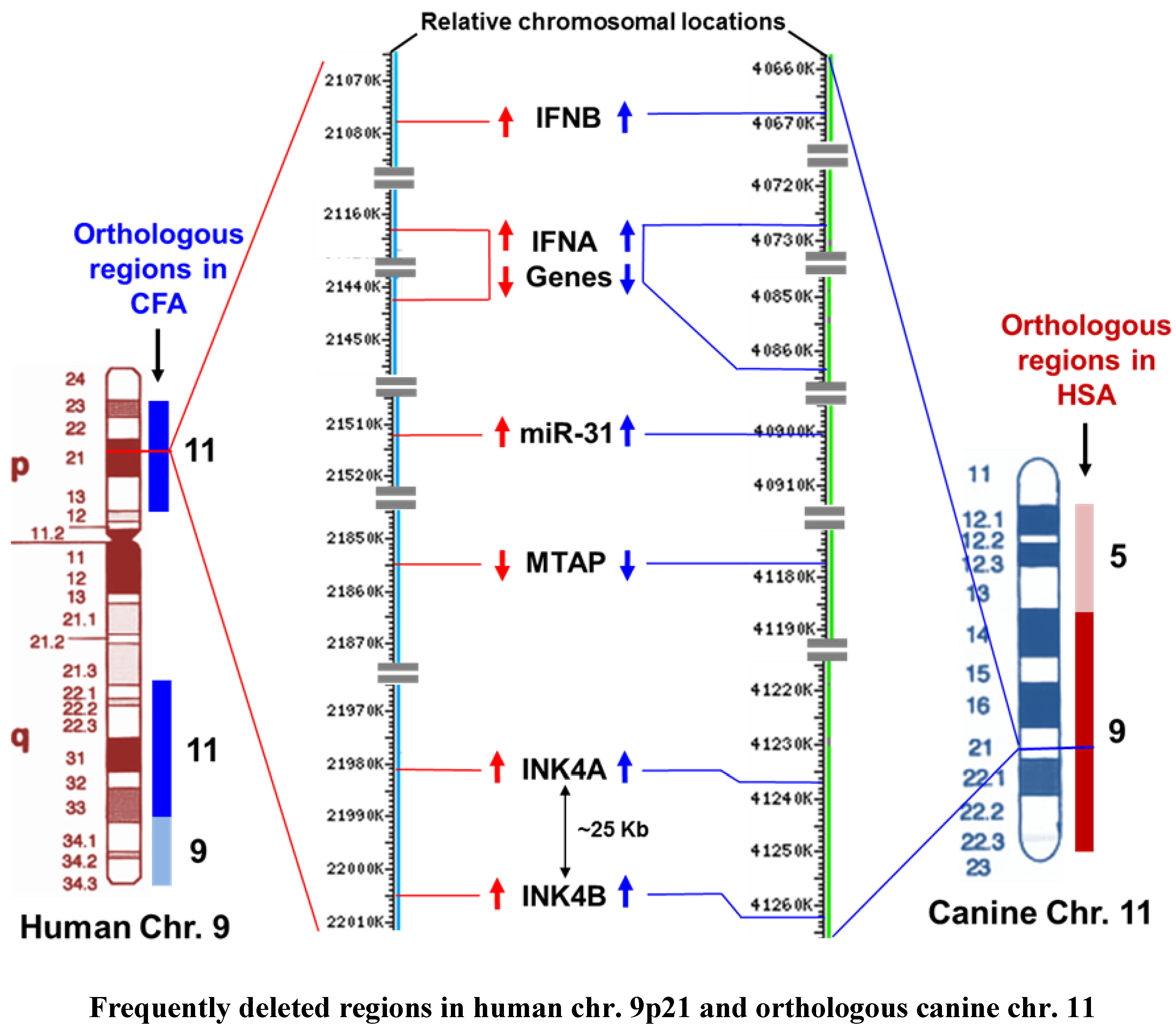
3.4. Alteration of the INK4A/ARF Locus in Human and Canine Cancers
4. Regulatory, Small Non-Coding RNAs: microRNAs in Cancers
4.1. OncomiRs: Cancer Associated miRNAs
4.2. Regulation of miRNAs in Human and Canine Breast Cancers
4.3. miRNAs Regulate Cell Cycle by Targeting Multiple Genes
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rowell, J.L.; McCarthy, D.O.; Alvarez, C.E. Dog models of naturally occurring cancer. Trends Mol. Med. 2011, 17, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.F.; Bird, R.C. Hematologic neoplasia—Gene therapy. In Schalm’s Veterinary Hematology; Weiss, D.J., Wardrop, K.J., Eds.; Wiley-Blackwell: Iowa City, IA, USA, 2010; pp. 550–557. [Google Scholar]
- American Veterinary Medical Association. Us Pet Ownership and Demographics Sourcebook 2007; American Veterinary Medical Association: Schaumburg, IL, USA, 2008. [Google Scholar]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Lindblad-Toh, K.; Vail, D.; London, C.; Bergman, P.; Barber, L.; Breen, M.; Kitchell, B.; McNeil, E.; Modiano, J.F.; et al. The dog as a cancer model. Nat. Biotechnol. 2006, 24, 1065–1066. [Google Scholar] [CrossRef] [PubMed]
- Vail, D.M.; MacEwen, E.G. Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Investig. 2000, 18, 781–792. [Google Scholar] [CrossRef]
- Cullen, J.M.; Page, R.; Misdorp, W. An overview of cancer pathogenesis, diagnosis and management. In Tumors in Domestic Animals; Moulton, D.J., Ed.; Blackwell Publishing Company, Iowa State Press: Ames, IA, USA, 2002; pp. 3–45. [Google Scholar]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer statistics, 2007. CA 2007, 57, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Owen, L.N. A comparative study of canine and human breast cancer. Investig. Cell Pathol. 1979, 2, 257–275. [Google Scholar]
- Sorenmo, K. Canine mammary gland tumors. Vet. Clin. North Am. Small Anim. Pract. 2003, 33, 573–596. [Google Scholar] [CrossRef]
- Ahern, T.E.; Bird, R.C.; Bird, A.E.; Wolfe, L.G. Expression of the oncogene c-erbb-2 in canine mammary cancers and tumor-derived cell lines. Am. J. Vet. Res. 1996, 57, 693–696. [Google Scholar] [PubMed]
- Misdorp, W. Tumors of the mammary gland. In Tumors in Domestic Animals; Meuten, D.J., Ed.; Iowa State Press: Ames, IA, USA, 2002; pp. 575–606. [Google Scholar]
- Sleeckx, N.; de Rooster, H.; Veldhuis Kroeze, E.J.; Van Ginneken, C.; Van Brantegem, L. Canine mammary tumours, an overview. Reprod. Domest. Anim. 2011, 46, 1112–1131. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, H.; Legare, M.E.; Mason, G.L.; Berkbigler, J.L.; Afzali, M.F.; Flint, A.F.; Hanneman, W.H. Significance of eralpha, her2, and cav1 expression and molecular subtype classification to canine mammary gland tumor. J. Vet. Diagn. Investig. 2014, 26, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Gama, A.; Alves, A.; Schmitt, F. Identification of molecular phenotypes in canine mammary carcinomas with clinical implications: Application of the human classification. Virchows Arch. 2008, 453, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Hennecke, S.; Bornemann-Kolatzki, K.; Urnovitz, H.B.; Neumann, S.; Strobel, P.; Kaup, F.J.; Brenig, B.; Schutz, E. Genome aberrations in canine mammary carcinomas and their detection in cell-free plasma DNA. PLoS ONE 2013, 8, e75485. [Google Scholar] [CrossRef] [PubMed]
- Sassi, F.; Benazzi, C.; Castellani, G.; Sarli, G. Molecular-based tumour subtypes of canine mammary carcinomas assessed by immunohistochemistry. BMC Vet. Res. 2010, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Lutful Kabir, F.M.; DeInnocentes, P.; Agarwal, P.; Riese, D.J.; Bird, R.C. Estrogen receptor-alpha, progesterone receptor and c-erbb/her-family receptor mrna detection and phenotype analysis in spontaneous canine models of breast cancer. Vet. Pathol. 2015. in preparation. [Google Scholar]
- Uva, P.; Aurisicchio, L.; Watters, J.; Loboda, A.; Kulkarni, A.; Castle, J.; Palombo, F.; Viti, V.; Mesiti, G.; Zappulli, V.; et al. Comparative expression pathway analysis of human and canine mammary tumors. BMC Genom. 2009, 10, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, N.A.; van Wolferen, M.E.; van den Ham, R.; van Leenen, D.; Groot Koerkamp, M.J.; Holstege, F.C.; Mol, J.A. Cdna microarray profiles of canine mammary tumour cell lines reveal deregulated pathways pertaining to their phenotype. Anim. Genet. 2008, 39, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Lutful Kabir, F.M.; Agarwal, P.; Deinnocentes, P.; Zaman, J.; Bird, A.C.; Bird, R.C. Novel frameshift mutation in the p16/ink4a tumor suppressor gene in canine breast cancer alters expression from the p16/ink4a/p14arf locus. J. Cell. Biochem. 2013, 114, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Klopfleisch, R.; Gruber, A.D. Differential expression of cell cycle regulators p21, p27 and p53 in metastasizing canine mammary adenocarcinomas versus normal mammary glands. Res. Vet. Sci. 2009, 87, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Van ‘t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Saal, L.H.; Johansson, P.; Holm, K.; Gruvberger-Saal, S.K.; She, Q.B.; Maurer, M.; Koujak, S.; Ferrando, A.A.; Malmstrom, P.; Memeo, L.; et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant pten tumor suppressor pathway activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7564–7569. [Google Scholar] [CrossRef] [PubMed]
- Sweet-Cordero, A.; Mukherjee, S.; Subramanian, A.; You, H.; Roix, J.J.; Ladd-Acosta, C.; Mesirov, J.; Golub, T.R.; Jacks, T. An oncogenic kras2 expression signature identified by cross-species gene-expression analysis. Nat. Genet. 2005, 37, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pines, J.; Hunter, T. Cyclin-dependent kinases: A new cell cycle motif? Trends Cell Biol. 1991, 1, 117–121. [Google Scholar] [CrossRef]
- Mareel, M.; Leroy, A. Clinical, cellular, and molecular aspects of cancer invasion. Physiol. Rev. 2003, 83, 337–376. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, A.; Demetrick, D.J.; Spillare, E.A.; Hagiwara, K.; Hussain, S.P.; Bennett, W.P.; Forrester, K.; Gerwin, B.; Serrano, M.; Beach, D.H.; et al. Mutations and altered expression of p16ink4 in human cancer. Proc. Natl. Acad. Sci. USA 1994, 91, 11045–11049. [Google Scholar] [CrossRef] [PubMed]
- Otterson, G.A.; Kratzke, R.A.; Coxon, A.; Kim, Y.W.; Kaye, F.J. Absence of p16ink4 protein is restricted to the subset of lung cancer lines that retains wildtype rb. Oncogene 1994, 9, 3375–3378. [Google Scholar] [PubMed]
- Harper, J.W.; Elledge, S.J. Cdk inhibitors in development and cancer. Curr. Opin. Genet. Dev. 1996, 6, 56–64. [Google Scholar] [CrossRef]
- Vidal, A.; Koff, A. Cell-cycle inhibitors: Three families united by a common cause. Gene 2000, 247, 1–15. [Google Scholar] [CrossRef]
- McConnell, B.B.; Gregory, F.J.; Stott, F.J.; Hara, E.; Peters, G. Induced expression of p16(ink4a) inhibits both cdk4- and cdk2-associated kinase activity by reassortment of cyclin-cdk-inhibitor complexes. Mol. Cell. Biol. 1999, 19, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Mahony, D.; Wills, K.; Lees, E. Cyclin d-cdk subunit arrangement is dependent on the availability of competing ink4 and p21 class inhibitors. Mol. Cell. Biol. 1999, 19, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Ruas, M.; Peters, G. The p16ink4a/cdkn2a tumor suppressor and its relatives. Biochim. Biophys. Acta 1998, 1378, F115–F177. [Google Scholar] [CrossRef]
- Sharpless, N.E. Ink4a/arf: A multifunctional tumor suppressor locus. Mutat. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; DePinho, R.A. The ink4a/arf locus and its two gene products. Curr. Opin. Genet. Dev. 1999, 9, 22–30. [Google Scholar] [CrossRef]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. P21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T.; Pines, J. Cyclins and cancer. Ii: Cyclin d and cdk inhibitors come of age. Cell 1994, 79, 573–582. [Google Scholar] [CrossRef]
- Kamb, A.; Gruis, N.A.; Weaver-Feldhaus, J.; Liu, Q.; Harshman, K.; Tavtigian, S.V.; Stockert, E.; Day, R.S., 3rd; Johnson, B.E.; Skolnick, M.H. A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Wesierska-Gadek, J.; Schmid, G. Dual action of the inhibitors of cyclin-dependent kinases: Targeting of the cell-cycle progression and activation of wild-type p53 protein. Expert Opin. Investig. Drugs 2006, 15, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M. The ink4a/arf locus in murine tumorigenesis. Carcinogenesis 2000, 21, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Hernandez, J.; Milne, B.S.; Queen, C.; O’Brien, P.C.; Hoather, T.; Haugland, S.; Ferguson-Smith, M.A.; Dobson, J.M.; Sargan, D.R. Disruption of chromosome 11 in canine fibrosarcomas highlights an unusual variability of cdkn2b in dogs. BMC Vet. Res. 2009, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- DeInnocentes, P.; Agarwal, P.; Bird, R.C. Phenotype-rescue of cyclin-dependent kinase inhibitor p16/ink4a defects in a spontaneous canine cell model of breast cancer. J. Cell. Biochem. 2009, 106, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Bianco, S.R.; Fosmire, S.; Wojcieszyn, J.; Modiano, J.F. Expression and significance of p53, rb, p21/waf-1, p16/ink-4a, and pten tumor suppressors in canine melanoma. Vet. Pathol. 2002, 39, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Migone, F.; Deinnocentes, P.; Smith, B.F.; Bird, R.C. Alterations in cdk1 expression and nuclear/nucleolar localization following induction in a spontaneous canine mammary cancer model. J. Cell. Biochem. 2006, 98, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Gilley, J.; Fried, M. One ink4 gene and no arf at the fugu equivalent of the human ink4a/arf/ink4b tumour suppressor locus. Oncogene 2001, 20, 7447–7452. [Google Scholar] [CrossRef] [PubMed]
- Byeon, I.J.; Li, J.; Ericson, K.; Selby, T.L.; Tevelev, A.; Kim, H.J.; O’Maille, P.; Tsai, M.D. Tumor suppressor p16ink4a: Determination of solution structure and analyses of its interaction with cyclin-dependent kinase 4. Mol. Cell 1998, 1, 421–431. [Google Scholar] [CrossRef]
- Venkataramani, R.; Swaminathan, K.; Marmorstein, R. Crystal structure of the cdk4/6 inhibitory protein p18ink4c provides insights into ankyrin-like repeat structure/function and tumor-derived p16ink4 mutations. Nat. Struct. Biol. 1998, 5, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Merlo, A.; Bedi, G.; Shapiro, G.I.; Edwards, C.D.; Rollins, B.J.; Sidransky, D. A novel p16ink4a transcript. Cancer Res. 1995, 55, 2995–2997. [Google Scholar] [PubMed]
- Guan, K.L.; Jenkins, C.W.; Li, Y.; O’Keefe, C.L.; Noh, S.; Wu, X.; Zariwala, M.; Matera, A.G.; Xiong, Y. Isolation and characterization of p19ink4d, a p16-related inhibitor specific to cdk6 and cdk4. Mol. Biol. Cell 1996, 7, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. G1 phase progression: Cycling on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Quelle, D.E.; Ashmun, R.A.; Hannon, G.J.; Rehberger, P.A.; Trono, D.; Richter, K.H.; Walker, C.; Beach, D.; Sherr, C.J.; Serrano, M. Cloning and characterization of murine p16ink4a and p15ink4b genes. Oncogene 1995, 11, 635–645. [Google Scholar] [PubMed]
- Chin, L.; Pomerantz, J.; Polsky, D.; Jacobson, M.; Cohen, C.; Cordon-Cardo, C.; Horner, J.W., 2nd; DePinho, R.A. Cooperative effects of ink4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997, 11, 2822–2834. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse ink4a locus mediated by the alternative reading frame product p19arf. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The ink4a tumor suppressor gene product, p19arf, interacts with mdm2 and neutralizes mdm2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef] [PubMed]
- Hussussian, C.J.; Struewing, J.P.; Goldstein, A.M.; Higgins, P.A.; Ally, D.S.; Sheahan, M.D.; Clark, W.H., Jr.; Tucker, M.A.; Dracopoli, N.C. Germline p16 mutations in familial melanoma. Nat. Genet. 1994, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kamb, A.; Shattuck-Eidens, D.; Eeles, R.; Liu, Q.; Gruis, N.A.; Ding, W.; Hussey, C.; Tran, T.; Miki, Y.; Weaver-Feldhaus, J.; et al. Analysis of the p16 gene (cdkn2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat. Genet. 1994, 8, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Ranade, K.; Hussussian, C.J.; Sikorski, R.S.; Varmus, H.E.; Goldstein, A.M.; Tucker, M.A.; Serrano, M.; Hannon, G.J.; Beach, D.; Dracopoli, N.C. Mutations associated with familial melanoma impair p16ink4 function. Nat. Genet. 1995, 10, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Caldas, C.; Hahn, S.A.; da Costa, L.T.; Redston, M.S.; Schutte, M.; Seymour, A.B.; Weinstein, C.L.; Hruban, R.H.; Yeo, C.J.; Kern, S.E. Frequent somatic mutations and homozygous deletions of the p16 (mts1) gene in pancreatic adenocarcinoma. Nat. Genet. 1994, 8, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Dreyling, M.H.; Bohlander, S.K.; Adeyanju, M.O.; Olopade, O.I. Detection of cdkn2 deletions in tumor cell lines and primary glioma by interphase fluorescence in situ hybridization. Cancer Res. 1995, 55, 984–988. [Google Scholar] [PubMed]
- Hatta, Y.; Hirama, T.; Miller, C.W.; Yamada, Y.; Tomonaga, M.; Koeffler, H.P. Homozygous deletions of the p15 (mts2) and p16 (cdkn2/mts1) genes in adult t-cell leukemia. Blood 1995, 85, 2699–2704. [Google Scholar] [PubMed]
- Nobori, T.; Miura, K.; Wu, D.J.; Lois, A.; Takabayashi, K.; Carson, D.A. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 1994, 368, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Hirano, N.; Sato, N.; Takahashi, T.; Hangaishi, A.; Tanaka, K.; Kurokawa, M.; Tanaka, T.; Mitani, K.; Yazaki, Y.; et al. Homozygous loss of the cyclin-dependent kinase 4-inhibitor (p16) gene in human leukemias. Blood 1994, 84, 2431–2435. [Google Scholar] [PubMed]
- Quesnel, B.; Preudhomme, C.; Philippe, N.; Vanrumbeke, M.; Dervite, I.; Lai, J.L.; Bauters, F.; Wattel, E.; Fenaux, P. P16 gene homozygous deletions in acute lymphoblastic leukemia. Blood 1995, 85, 657–663. [Google Scholar] [PubMed]
- Carrera, C.J.; Eddy, R.L.; Shows, T.B.; Carson, D.A. Assignment of the gene for methylthioadenosine phosphorylase to human chromosome 9 by mouse-human somatic cell hybridization. Proc. Natl. Acad. Sci. USA 1984, 81, 2665–2668. [Google Scholar] [CrossRef] [PubMed]
- Nobori, T.; Takabayashi, K.; Tran, P.; Orvis, L.; Batova, A.; Yu, A.L.; Carson, D.A. Genomic cloning of methylthioadenosine phosphorylase: A purine metabolic enzyme deficient in multiple different cancers. Proc. Natl. Acad. Sci. USA 1996, 93, 6203–6208. [Google Scholar] [CrossRef] [PubMed]
- Herzog, C.R.; Noh, S.; Lantry, L.E.; Guan, K.L.; You, M. Cdkn2a encodes functional variation of p16ink4a but not p19arf, which confers selection in mouse lung tumorigenesis. Mol. Carcinog. 1999, 25, 92–98. [Google Scholar] [CrossRef]
- Zhang, S.; Ramsay, E.S.; Mock, B.A. Cdkn2a, the cyclin-dependent kinase inhibitor encoding p16ink4a and p19arf, is a candidate for the plasmacytoma susceptibility locus, pctr1. Proc. Natl. Acad. Sci. USA 1998, 95, 2429–2434. [Google Scholar] [CrossRef] [PubMed]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lee, H.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R.A. Role of the ink4a locus in tumor suppression and cell mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R.A. Loss of p16ink4a with retention of p19arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.A.; Fleischli, M.A. Inactivation of p53 and retinoblastoma family pathways in canine osteosarcoma cell lines. Vet. Pathol. 2000, 37, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Yeudall, W.A.; Crawford, R.Y.; Ensley, J.F.; Robbins, K.C. Mts1/cdk4i is altered in cell lines derived from primary and metastatic oral squamous cell carcinoma. Carcinogenesis 1994, 15, 2683–2686. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; DuBois, W.; Ramsay, E.S.; Bliskovski, V.; Morse, H.C., 3rd; Taddesse-Heath, L.; Vass, W.C.; DePinho, R.A.; Mock, B.A. Efficiency alleles of the pctr1 modifier locus for plasmacytoma susceptibility. Mol. Cell. Biol. 2001, 21, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Shearin, A.L.; Hedan, B.; Cadieu, E.; Erich, S.A.; Schmidt, E.V.; Faden, D.L.; Cullen, J.; Abadie, J.; Kwon, E.M.; Grone, A.; et al. The mtap-cdkn2a locus confers susceptibility to a naturally occurring canine cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Melkamu, T.; Zhang, X.; Tan, J.; Zeng, Y.; Kassie, F. Alteration of microrna expression in vinyl carbamate-induced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol. Carcinogenesis 2010, 31, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the encode pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.F. Non-coding rnas: Meet thy masters. BioEssays: News Rev. Mol. Cell. Dev. Biol. 2010, 32, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chinnaiyan, A.M. The emergence of lncrnas in cancer biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding rnas and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding rna in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Sunkin, S.M.; Mehler, M.F.; Mattick, J.S. Specific expression of long noncoding rnas in the mouse brain. Proc. Natl. Acad. Sci. USA 2008, 105, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding rna hotair reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding rna as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jones, A.; Sun, C.W.; Li, C.; Chang, C.W.; Joo, H.Y.; Dai, Q.; Mysliwiec, M.R.; Wu, L.C.; Guo, Y.; et al. Prc2 complexes with jarid2, mtf2, and esprc2p48 in es cells to modulate es cell pluripotency and somatic cell reprogramming. Stem Cells 2011, 29, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Hengartner, M.O. Mirnas and apoptosis: Rnas to die for. Oncogene 2006, 25, 6176–6187. [Google Scholar] [CrossRef] [PubMed]
- Schickel, R.; Boyerinas, B.; Park, S.M.; Peter, M.E. Micrornas: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008, 27, 5959–5974. [Google Scholar] [CrossRef] [PubMed]
- Stefani, G.; Slack, F.J. Small non-coding rnas in animal development. Nat. Rev. Mol. Cell Biol. 2008, 9, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed rnas. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—Micrornas with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microrna genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Calin, G.A.; Croce, C.M. Micrornas in cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mrna translation and stability by micrornas. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mrnas are conserved targets of micrornas. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Kent, O.A.; Mendell, J.T. A small piece in the cancer puzzle: Micrornas as tumor suppressors and oncogenes. Oncogene 2006, 25, 6188–6196. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Kashima, R. Dysregulation of microRNA biogenesis machinery in cancer. Crit. Rev. Biochem. Molec. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. Trbp recruits the dicer complex to ago2 for microrna processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An rna-directed nuclease mediates post-transcriptional gene silencing in drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [PubMed]
- Zeng, Y.; Wagner, E.J.; Cullen, B.R. Both natural and designed micro rnas can inhibit the expression of cognate mrnas when expressed in human cells. Mol. Cell 2002, 9, 1327–1333. [Google Scholar] [CrossRef]
- Martello, G.; Rosato, A.; Ferrari, F.; Manfrin, A.; Cordenonsi, M.; Dupont, S.; Enzo, E.; Guzzardo, V.; Rondina, M.; Spruce, T.; et al. A microrna targeting dicer for metastasis control. Cell 2010, 141, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- rna genes mir15 and mir16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of mir-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microrna-127 with downregulation of the proto-oncogene bcl6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A microrna signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. Microrna gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. Microrna expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microrna expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microrna molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Kluiver, J.; Poppema, S.; de Jong, D.; Blokzijl, T.; Harms, G.; Jacobs, S.; Kroesen, B.J.; van den Berg, A. Bic and mir-155 are highly expressed in hodgkin, primary mediastinal and diffuse large b cell lymphomas. J. Pathol. 2005, 207, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Metzler, M.; Wilda, M.; Busch, K.; Viehmann, S.; Borkhardt, A. High expression of precursor microrna-155/bic rna in children with burkitt lymphoma. Genes Chromosomes Cancer 2004, 39, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Ciafre, S.A.; Galardi, S.; Mangiola, A.; Ferracin, M.; Liu, C.G.; Sabatino, G.; Negrini, M.; Maira, G.; Croce, C.M.; Farace, M.G. Extensive modulation of a set of micrornas in primary glioblastoma. Biochem. Biophys. Res. Commun. 2005, 334, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Garofalo, M.; Martelli, M.P.; Briesewitz, R.; Wang, L.; Fernandez-Cymering, C.; Volinia, S.; Liu, C.G.; Schnittger, S.; Haferlach, T.; et al. Distinctive microrna signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc. Natl. Acad. Sci. USA 2008, 105, 3945–3950. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. Microrna-21 regulates expression of the pten tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. Microrna-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A.H. Programmed cell death 4 (pdcd4) is an important functional target of the microrna mir-21 in breast cancer cells. J. Biol. Chem. 2008, 283, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Si, M.L.; Wu, H.; Mo, Y.Y. Microrna-21 targets the tumor suppressor gene tropomyosin 1 (tpm1). J. Biol. Chem. 2007, 282, 14328–14336. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. Ras is regulated by the let-7 microrna family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Nakagawa, Y.; Naoe, T. Let-7 microrna functions as a potential growth suppressor in human colon cancer cells. Biol. Pharm. Bull. 2006, 29, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Dutta, A. The tumor suppressor microrna let-7 represses the hmga2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. Microrna let-7a down-regulates myc and reverts myc-induced growth in burkitt lymphoma cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar] [CrossRef] [PubMed]
- Harquail, J.; Benzina, S.; Robichaud, G.A. Micrornas and breast cancer malignancy: An overview of mirna-regulated cancer processes leading to metastasis. Cancer Biomark.: Sect. Dis. Markers 2012, 11, 269–280. [Google Scholar]
- Zhang, Z.J.; Ma, S.L. Mirnas in breast cancer tumorigenesis (review). Oncol. Rep. 2012, 27, 903–910. [Google Scholar] [PubMed]
- Qi, L.; Bart, J.; Tan, L.P.; Platteel, I.; Sluis, T.; Huitema, S.; Harms, G.; Fu, L.; Hollema, H.; Berg, A. Expression of mir-21 and its targets (pten, pdcd4, tm1) in flat epithelial atypia of the breast in relation to ductal carcinoma in situ and invasive carcinoma. BMC Cancer 2009, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wu, H.; Wu, F.; Nie, D.; Sheng, S.; Mo, Y.Y. Microrna-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008, 18, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Boggs, R.M.; Wright, Z.M.; Stickney, M.J.; Porter, W.W.; Murphy, K.E. Microrna expression in canine mammary cancer. Mamm. Genome 2008, 19, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Schelter, J.; Burchard, J.; Kibukawa, M.; Martin, M.M.; Bartz, S.R.; Johnson, J.M.; Cummins, J.M.; Raymond, C.K.; Dai, H.; et al. Transcripts targeted by the microrna-16 family cooperatively regulate cell cycle progression. Mol. Cell. Biol. 2007, 27, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fu, H.; Sun, F.; Zhang, H.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Mir-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008, 36, 5391–5404. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, F.; Patrawala, L.; Osaki, M.; Takahashi, R.U.; Yamamoto, Y.; Kosaka, N.; Kawamata, M.; Kelnar, K.; Bader, A.G.; Brown, D.; et al. Systemic delivery of synthetic microrna-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol. Ther. 2010, 18, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Fu, X.D.; Zhou, Y.; Zhang, Y. Down-regulation of the cyclin e1 oncogene expression by microrna-16–1 induces cell cycle arrest in human cancer cells. BMB Rep. 2009, 42, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.J.; Malumbres, M. Micrornas and the cell cycle. Biochim. Biophys. Acta 2011, 1812, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Fu, H.; Liu, Q.; Tie, Y.; Zhu, J.; Xing, R.; Sun, Z.; Zheng, X. Downregulation of ccnd1 and cdk6 by mir-34a induces cell cycle arrest. FEBS Lett. 2008, 582, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microrna component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Ropero, S.; Ballestar, E.; Fraga, M.F.; Cerrato, C.; Setien, F.; Casado, S.; Suarez-Gauthier, A.; Sanchez-Cespedes, M.; Git, A.; et al. Genetic unmasking of an epigenetically silenced microrna in human cancer cells. Cancer Res. 2007, 67, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Blelloch, R. Cell cycle regulation by micrornas in embryonic stem cells. Cancer Res. 2009, 69, 4093–4096. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Kim, H.H.; Abdelmohsen, K.; Kuwano, Y.; Pullmann, R., Jr.; Srikantan, S.; Subrahmanyam, R.; Martindale, J.L.; Yang, X.; Ahmed, F.; et al. P16(ink4a) translation suppressed by mir-24. PLoS ONE 2008, 3, e1864. [Google Scholar] [CrossRef] [PubMed]
- Malhas, A.; Saunders, N.J.; Vaux, D.J. The nuclear envelope can control gene expression and cell cycle progression via mirna regulation. Cell Cycle 2010, 9, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zou, F.; Zhang, X.; Li, H.; Dulak, A.; Tomko, R.J., Jr.; Lazo, J.S.; Wang, Z.; Zhang, L.; Yu, J. Microrna-21 negatively regulates cdc25a and cell cycle progression in colon cancer cells. Cancer Res. 2009, 69, 8157–8165. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Dey, B.K.; Dutta, A. Mir-322/424 and -503 are induced during muscle differentiation and promote cell cycle quiescence and differentiation by down-regulation of cdc25a. Mol. Biol. Cell 2010, 21, 2138–2149. [Google Scholar] [CrossRef] [PubMed]
- Kabir, F.M.L.; DeInnocentes, P.; Bird, R.C. Altered microrna expression profiles and regulation of ink4a/cdkn2a tumor suppressor genes in canine breast cancer models. J. Cell. Biochem. 2015, in press. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lutful Kabir, F.M.; Alvarez, C.E.; Bird, R.C. Canine Mammary Carcinomas: A Comparative Analysis of Altered Gene Expression. Vet. Sci. 2016, 3, 1. https://doi.org/10.3390/vetsci3010001
Lutful Kabir FM, Alvarez CE, Bird RC. Canine Mammary Carcinomas: A Comparative Analysis of Altered Gene Expression. Veterinary Sciences. 2016; 3(1):1. https://doi.org/10.3390/vetsci3010001
Chicago/Turabian StyleLutful Kabir, Farruk M., Carlos E. Alvarez, and R. Curtis Bird. 2016. "Canine Mammary Carcinomas: A Comparative Analysis of Altered Gene Expression" Veterinary Sciences 3, no. 1: 1. https://doi.org/10.3390/vetsci3010001
APA StyleLutful Kabir, F. M., Alvarez, C. E., & Bird, R. C. (2016). Canine Mammary Carcinomas: A Comparative Analysis of Altered Gene Expression. Veterinary Sciences, 3(1), 1. https://doi.org/10.3390/vetsci3010001