
The Kitty Microbiome Project: Defining the Healthy Fecal “Core Microbiome” in Pet Domestic Cats
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Participatory Research
2.2. Sample and Metadata Collection
2.3. Sample Processing and Sequencing
2.4. Sequence Data Processing
2.5. Core Microbiome of Healthy Cats
2.6. Effect of Age on the Microbiome
2.7. Effect of Diet on the Microbiome
2.8. Effect of FIV Status and Living Environment on the Microbiome
3. Results
3.1. The Core Microbiome of Healthy Pet Cats
3.2. Effect of Age on the Microbiome
3.3. Effects of Diet on the Microbiome
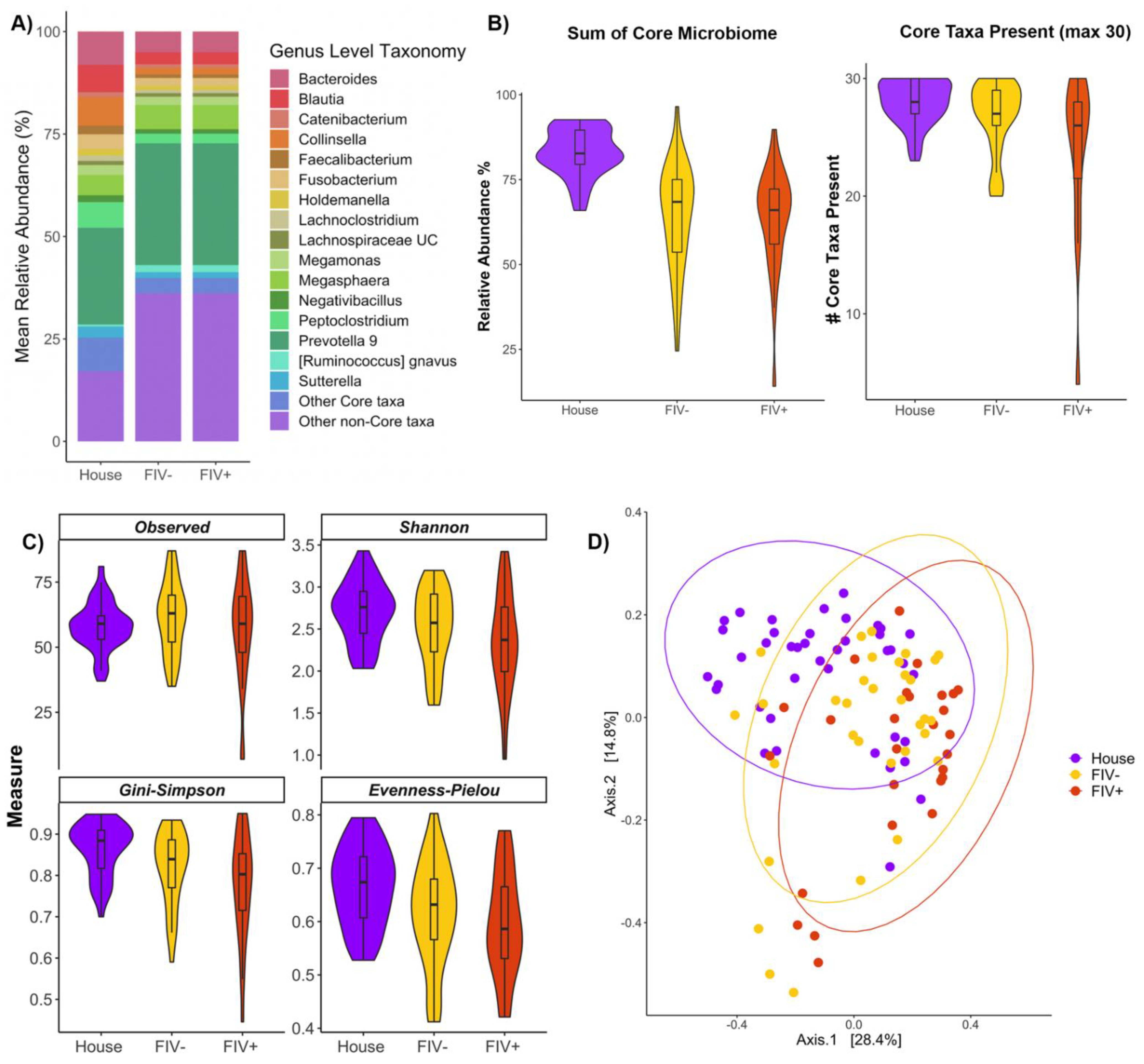
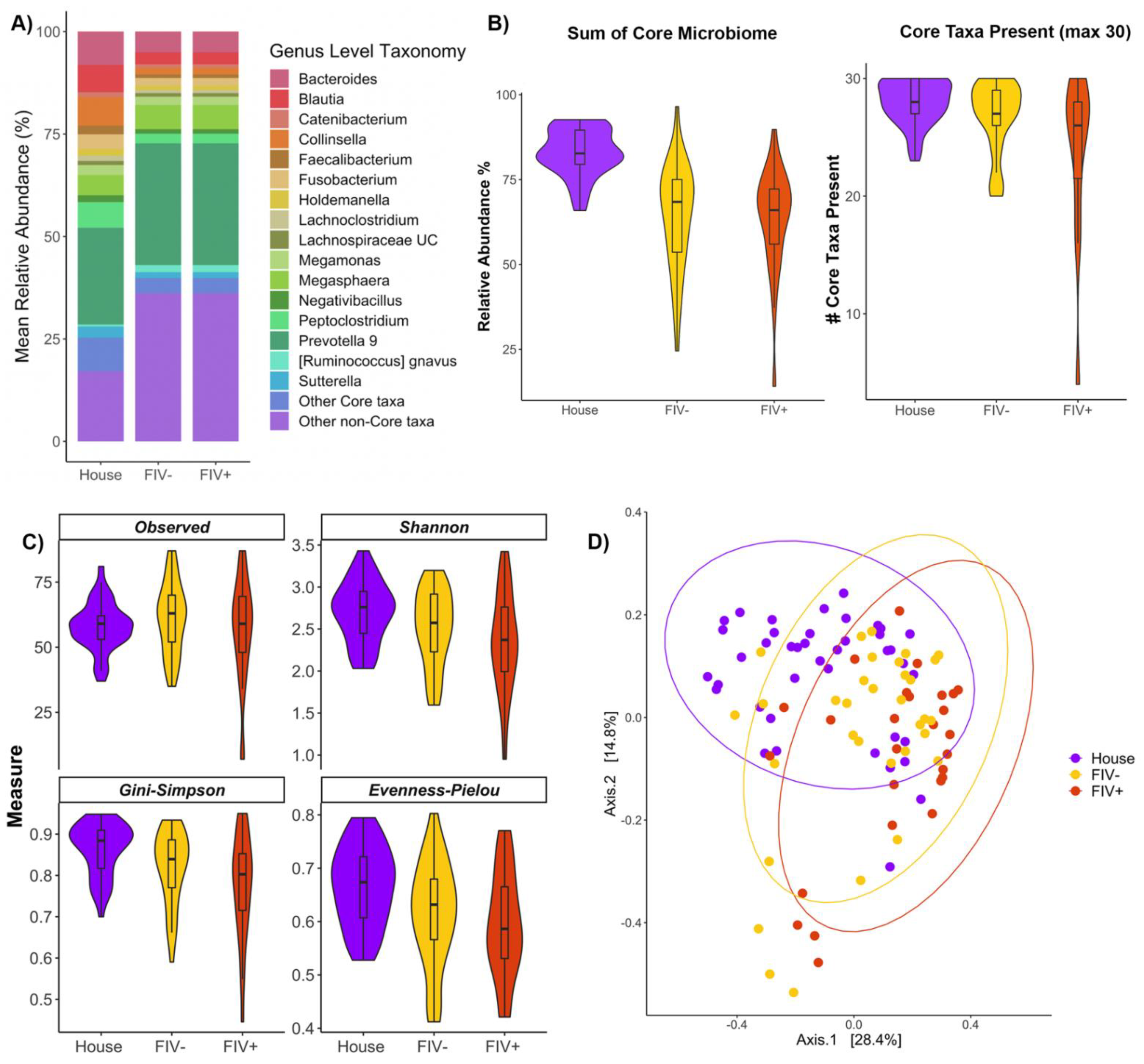
3.4. Effects of FIV Status and Living Environment on the Microbiome
4. Discussion
4.1. Core Microbiome
4.2. Age
4.3. Diet
4.4. Environment
4.5. FIV Status
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sikalidis, A.K.; Maykish, A. The Gut Microbiome and Type 2 Diabetes Mellitus: Discussing a Complex Relationship. Biomedicines 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallianou, N.; Stratigou, T.; Christodoulatos, G.S.; Dalamaga, M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Obesity and Obesity-Associated Metabolic Disorders: Current Evidence and Perspectives. Curr. Obes. Rep. 2019, 8, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Lourenςo, T.G.B.; Spencer, S.J.; Alm, E.J.; Colombo, A.P.V. Defining the Gut Microbiota in Individuals with Periodontal Diseases: An Exploratory Study. J. Oral Microbiol. 2018, 10, 1487741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimentel, M.; Lembo, A. Microbiome and Its Role in Irritable Bowel Syndrome. Dig. Dis. Sci. 2020, 65, 829–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, I.; Ramser, A.; Isham, N.; Ghannoum, M.A. The Gut Microbiome as a Major Regulator of the Gut-Skin Axis. Front. Microbiol. 2018, 9, 1459. [Google Scholar] [CrossRef] [Green Version]
- Jalanka-Tuovinen, J.; Salonen, A.; Nikkilä, J.; Immonen, O.; Kekkonen, R.; Lahti, L.; Palva, A.; de Vos, W.M. Intestinal Microbiota in Healthy Adults: Temporal Analysis Reveals Individual and Common Core and Relation to Intestinal Symptoms. PLoS ONE 2011, 6, e23035. [Google Scholar] [CrossRef]
- Marsilio, S.; Pilla, R.; Sarawichitr, B.; Chow, B.; Hill, S.L.; Ackermann, M.R.; Estep, J.S.; Lidbury, J.A.; Steiner, J.M.; Suchodolski, J.S. Characterization of the Fecal Microbiome in Cats with Inflammatory Bowel Disease or Alimentary Small Cell Lymphoma. Sci. Rep. 2019, 9, 19208. [Google Scholar] [CrossRef] [Green Version]
- Kieler, I.N.; Osto, M.; Hugentobler, L.; Puetz, L.; Gilbert, M.T.P.; Hansen, T.; Pedersen, O.; Reusch, C.E.; Zini, E.; Lutz, T.A.; et al. Diabetic Cats Have Decreased Gut Microbial Diversity and a Lack of Butyrate Producing Bacteria. Sci. Rep. 2019, 9, 4822. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.X.; Bicalho, R.C.; Fiani, N.; Lima, S.F.; Peralta, S. The Subgingival Microbial Community of Feline Periodontitis and Gingivostomatitis: Characterization and Comparison between Diseased and Healthy Cats. Sci. Rep. 2019, 9, 12340. [Google Scholar] [CrossRef] [Green Version]
- Older, C.E.; Diesel, A.; Patterson, A.P.; Meason-Smith, C.; Johnson, T.J.; Mansell, J.; Suchodolski, J.S.; Rodrigues Hoffmann, A. The Feline Skin Microbiota: The Bacteria Inhabiting the Skin of Healthy and Allergic Cats. PLoS ONE 2017, 12, e0178555. [Google Scholar] [CrossRef]
- Marsilio, S. Feline Chronic Enteropathy. J. Small Anim. Pract. 2021, 62, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, F.; Ghosh, T.S.; O’Toole, P.W. The Healthy Microbiome-What Is the Definition of a Healthy Gut Microbiome? Gastroenterology 2021, 160, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Fraser, C.M.; Ringel, Y.; Sanders, M.E.; Sartor, R.B.; Sherman, P.M.; Versalovic, J.; Young, V.; Finlay, B.B. Defining a Healthy Human Gut Microbiome: Current Concepts, Future Directions, and Clinical Applications. Cell Host Microbe 2012, 12, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The Healthy Human Microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Hyde, E.; Debelius, J.W.; Morton, J.T.; Gonzalez, A.; Ackermann, G.; Aksenov, A.A.; Behsaz, B.; Brennan, C.; Chen, Y.; et al. American Gut: An Open Platform for Citizen Science Microbiome Research. mSystems 2018, 3, e00031-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef] [Green Version]
- McBurney, M.I.; Davis, C.; Fraser, C.M.; Schneeman, B.O.; Huttenhower, C.; Verbeke, K.; Walter, J.; Latulippe, M.E. Establishing What Constitutes a Healthy Human Gut Microbiome: State of the Science, Regulatory Considerations, and Future Directions. J. Nutr. 2019, 149, 1882–1895. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lang, T.; Shen, J.; Dai, J.; Tian, L.; Wang, X. Core Gut Bacteria Analysis of Healthy Mice. Front. Microbiol. 2019, 10, 887. [Google Scholar] [CrossRef]
- You, I.; Kim, M.J. Comparison of Gut Microbiota of 96 Healthy Dogs by Individual Traits: Breed, Age, and Body Condition Score. Animals 2021, 11, 2432. [Google Scholar] [CrossRef]
- Dahlhausen, K.; Krebs, B.L.; Watters, J.V.; Ganz, H.H. Crowdfunding Campaigns Help Researchers Launch Projects and Generate Outreach. J. Microbiol. Biol. Educ. 2016, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A Communal Catalogue Reveals Earth’s Multiscale Microbial Diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler, M.; Coskun, Ö.K.; Ortega-Arbulú, A.-S.; Conci, N.; Wörheide, G.; Vargas, S.; Orsi, W.D. A 16S rRNA Gene Sequencing and Analysis Protocol for the Illumina MiniSeq Platform. Microbiologyopen 2018, 7, e00611. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-Species Living Tree Project (LTP)” Taxonomic Frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.r-project.org/ (accessed on 30 September 2022).
- Maechler, M.; Ringach, D. Diptest: Hartigan’s Dip Test Statistic for Unimodality. R Package, Version 0.75–5. 2013. Available online: https://CRAN.R-project.org/package=diptest (accessed on 30 September 2022).
- Lahti, L.; Shetty, S.; Blake, T.; Salojarvi, J. Tools for Microbiome Analysis in R. R Package, Version 1.14.0. 2017. Available online: https://cran.r-project.org/web/packages/metamicrobiomeR/index.html (accessed on 30 September 2022).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.A.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. 2021. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 30 September 2022).
- Salazar, G. EcolUtils: Utilities for Community Ecology Analysis; R Package Version 0.1. 2022. Available online: https://github.com/GuillemSalazar/EcolUtils (accessed on 30 September 2022).
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-Scale Meta-Omics Studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Dillon, M.R.; Bolyen, E.; Kaehler, B.D.; Huttley, G.A.; Caporaso, J.G. q2-Sample-Classifier: Machine-Learning Tools for Microbiome Classification and Regression. J. Open Res. Softw. 2018, 3, 934. [Google Scholar] [CrossRef]
- Alessandri, G.; Milani, C.; Mancabelli, L.; Longhi, G.; Anzalone, R.; Lugli, G.A.; Duranti, S.; Turroni, F.; Ossiprandi, M.C.; van Sinderen, D.; et al. Deciphering the Bifidobacterial Populations within the Canine and Feline Gut Microbiota. Appl. Environ. Microbiol. 2020, 86, e02875-19. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.L.; Swift, N.C.; Forster-van Hijfte, M.; Rutgers, H.C.; Lamport, A.; Ballèvre, O.; Batt, R.M. Comparison of the Bacterial Flora of the Duodenum in Healthy Cats and Cats with Signs of Gastrointestinal Tract Disease. J. Am. Vet. Med. Assoc. 2001, 218, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Hooda, S.; Vester Boler, B.M.; Kerr, K.R.; Dowd, S.E.; Swanson, K.S. The Gut Microbiome of Kittens Is Affected by Dietary Protein:carbohydrate Ratio and Associated with Blood Metabolite and Hormone Concentrations. Br. J. Nutr. 2013, 109, 1637–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mead, G.C. The Amino Acid-Fermenting Clostridia. J. Gen. Microbiol. 1971, 67, 47–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butowski, C.F.; Thomas, D.G.; Young, W.; Cave, N.J.; McKenzie, C.M.; Rosendale, D.I.; Bermingham, E.N. Addition of Plant Dietary Fibre to a Raw Red Meat High Protein, High Fat Diet, Alters the Faecal Bacteriome and Organic Acid Profiles of the Domestic Cat (Felis Catus). PLoS ONE 2019, 14, e0216072. [Google Scholar] [CrossRef] [Green Version]
- Levine Uri, Y.; Torey, L.; Allen Heather, K.; Stanton Thad, B. Butyrate-Producing Bacteria, Including Mucin Degraders, from the Swine Intestinal Tract. Appl. Environ. Microbiol. 2013, 79, 3879–3881. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.L.; Yap, Y.A.; McLeod, K.H.; Mackay, C.R.; Mariño, E. Dietary Metabolites and the Gut Microbiota: An Alternative Approach to Control Inflammatory and Autoimmune Diseases. Clin. Transl. Immunol. 2016, 5, e82. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Formation of Propionate and Butyrate by the Human Colonic Microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-Fat-Diet-Induced Obesity in Mice by Regulating G Protein-Coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef]
- Sun, M.; Wu, W.; Liu, Z.; Cong, Y. Microbiota Metabolite Short Chain Fatty Acids, GPCR, and Inflammatory Bowel Diseases. J. Gastroenterol. 2017, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of Short Chain Fatty Acids by the Gut Microbiota and Their Impact on Human Metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deusch, O.; O’Flynn, C.; Colyer, A.; Swanson, K.S.; Allaway, D.; Morris, P. A Longitudinal Study of the Feline Faecal Microbiome Identifies Changes into Early Adulthood Irrespective of Sexual Development. PLoS ONE 2015, 10, e0144881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, R.; Wilms, E.; Masclee, A.A.M.; Smidt, H.; Zoetendal, E.G.; Jonkers, D. Age-Dependent Changes in GI Physiology and Microbiota: Time to Reconsider? Gut 2018, 67, 2213–2222. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, H.; Shimada, K.; Kiyosue-Yasuda, T.; Kiyosue, M.; Oishi, Y.; Kimura, S.; Ohashi, Y.; Fujisawa, T.; Hotta, K.; Yamada, A.; et al. Transition of the Intestinal Microbiota of Cats with Age. PLoS ONE 2017, 12, e0181739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermingham, E.N.; Young, W.; Butowski, C.F.; Moon, C.D.; Maclean, P.H.; Rosendale, D.; Cave, N.J.; Thomas, D.G. The Fecal Microbiota in the Domestic Cat (Felis Catus) Is Influenced by Interactions Between Age and Diet; A Five Year Longitudinal Study. Front. Microbiol. 2018, 9, 1231. [Google Scholar] [CrossRef]
- Wernimont, S.M.; Radosevich, J.; Jackson, M.I.; Ephraim, E.; Badri, D.V.; MacLeay, J.M.; Jewell, D.E.; Suchodolski, J.S. The Effects of Nutrition on the Gastrointestinal Microbiome of Cats and Dogs: Impact on Health and Disease. Front. Microbiol. 2020, 11, 1266. [Google Scholar] [CrossRef]
- Li, Q.; Pan, Y. Differential Responses to Dietary Protein and Carbohydrate Ratio on Gut Microbiome in Obese vs. Lean Cats. Front. Microbiol. 2020, 11, 591462. [Google Scholar] [CrossRef]
- Freeman, L.M.; Chandler, M.L.; Hamper, B.A.; Weeth, L.P. Current Knowledge about the Risks and Benefits of Raw Meat–based Diets for Dogs and Cats. J. Am. Vet. Med. Assoc. 2013, 243, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.-Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Schmidt, M.; Unterer, S.; Suchodolski, J.S.; Honneffer, J.B.; Guard, B.C.; Lidbury, J.A.; Steiner, J.M.; Fritz, J.; Kölle, P. The Fecal Microbiome and Metabolome Differs between Dogs Fed Bones and Raw Food (BARF) Diets and Dogs Fed Commercial Diets. PLoS ONE 2018, 13, e0201279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, R.A.; Warrior, J.R. Clicker Training Increases Exploratory Behaviour and Time Spent at the Front of the Enclosure in Shelter Cats; Implications for Welfare and Adoption Rates. Appl. Anim. Behav. Sci. 2019, 211, 77–83. [Google Scholar] [CrossRef]
- Wagner, D.; Hurley, K.; Stavisky, J. Shelter Housing for Cats: Practical Aspects of Design and Construction, and Adaptation of Existing Accommodation. J. Feline Med. Surg. 2018, 20, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Foreman-Worsley, R.; Farnworth, M.J. A Systematic Review of Social and Environmental Factors and Their Implications for Indoor Cat Welfare. Appl. Anim. Behav. Sci. 2019, 220, 104841. [Google Scholar] [CrossRef]
- Kates, A.E.; Jarrett, O.; Skarlupka, J.H.; Sethi, A.; Duster, M.; Watson, L.; Suen, G.; Poulsen, K.; Safdar, N. Household Pet Ownership and the Microbial Diversity of the Human Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zottler, E.-M.; Bieri, M.; Basso, W.; Schnyder, M. Intestinal Parasites and Lungworms in Stray, Shelter and Privately Owned Cats of Switzerland. Parasitol. Int. 2019, 69, 75–81. [Google Scholar] [CrossRef]
- Hoggard, K.R.; Jarriel, D.M.; Bevelock, T.J.; Verocai, G.G. Prevalence Survey of Gastrointestinal and Respiratory Parasites of Shelter Cats in Northeastern Georgia, USA. Vet. Parasitol. Reg. Stud. Rep. 2019, 16, 100270. [Google Scholar] [CrossRef]
- Willby, R. Afonso Respiratory Disease in the Cat in the Shelter Environment. In BSAVA Manual Canine and Feline Shelter Medicine; British Small Animal Veterinary Association: Gloucester, UK, 2018. [Google Scholar] [CrossRef]
- Šlapeta, J.; Dowd, S.E.; Alanazi, A.D.; Westman, M.E.; Brown, G.K. Differences in the Faecal Microbiome of Non-Diarrhoeic Clinically Healthy Dogs and Cats Associated with Giardia Duodenalis Infection: Impact of Hookworms and Coccidia. Int. J. Parasitol. 2015, 45, 585–594. [Google Scholar] [CrossRef]
- Vlčková, K.; Pafčo, B.; Petrželková, K.J.; Modrý, D.; Todd, A.; Yeoman, C.J.; Torralba, M.; Wilson, B.A.; Stumpf, R.M.; White, B.A.; et al. Relationships between Gastrointestinal Parasite Infections and the Fecal Microbiome in Free-Ranging Western Lowland Gorillas. Front. Microbiol. 2018, 9, 1202. [Google Scholar] [CrossRef]
- Ryan, G.; Grimes, T.; Brankin, B.; Mabruk, M.J.E.M.F.; Hosie, M.J.; Jarrett, O.; Callanan, J.J. Neuropathology Associated with Feline Immunodeficiency Virus Infection Highlights Prominent Lymphocyte Trafficking through Both the Blood-Brain and Blood-Choroid Plexus Barriers. J. Neurovirol. 2005, 11, 337–345. [Google Scholar] [CrossRef]
- Hartmann, K. Clinical Aspects of Feline Immunodeficiency and Feline Leukemia Virus Infection. Vet. Immunol. Immunopathol. 2011, 143, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Weese, J.S.; Nichols, J.; Jalali, M.; Litster, A. The Rectal Microbiota of Cats Infected with Feline Immunodeficiency Virus Infection and Uninfected Controls. Vet. Microbiol. 2015, 180, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Wasimuddin; Brändel, S.D.; Tschapka, M.; Page, R.; Rasche, A.; Corman, V.M.; Drosten, C.; Sommer, S. Astrovirus Infections Induce Age-Dependent Dysbiosis in Gut Microbiomes of Bats. ISME J. 2018, 12, 2883–2893. [Google Scholar] [CrossRef] [PubMed]
- Barbian, H.J.; Li, Y.; Ramirez, M.; Klase, Z.; Lipende, I.; Mjungu, D.; Moeller, A.H.; Wilson, M.L.; Pusey, A.E.; Lonsdorf, E.V.; et al. Destabilization of the Gut Microbiome Marks the End-Stage of Simian Immunodeficiency Virus Infection in Wild Chimpanzees. Am. J. Primatol. 2018, 80, 2515. [Google Scholar] [CrossRef]
- Wasimuddin; Corman, V.M.; Ganzhorn, J.U.; Rakotondranary, J.; Ratovonamana, Y.R.; Drosten, C.; Sommer, S. Adenovirus Infection Is Associated with Altered Gut Microbial Communities in a Non-Human Primate. Sci. Rep. 2019, 9, 13410. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Age Class | n | Age (Months) | Fecal Score | Body Condition Score | F (%) | SN (%) |
|---|---|---|---|---|---|---|
| Junior | 58 | 21.5 (±8.48) | 2.7 (±1.04) | 5.2 (±0.38) | 47 | 95 |
| Prime | 59 | 58.7 (±15.28) | 2.7 (±0.95) | 5 (±0.61) | 61 | 86 |
| Mature | 44 | 109.5 (±15.84) | 3.1 (±0.99) | 5.1 (±0.56) | 59 | 93 |
| Senior | 6 | 141.3 (±4.32) | 3.2 (±0.5) | 5.6 (±0.55) | 50 | 83 |
| Phylum | Class | Order | Family | Genus | Median | SD | Prev. | 2.5th | 10th | 90th | 97.5th |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Actinobacteria | Coriobacteriia | Coriobacteriales | Coriobacteriaceae | Collinsella | 5.93 | 6.456 | 96.4 | 0.81 | 1.62 | 16.98 | 25.35 |
| Actinobacteria | Coriobacteriia | Coriobacteriales | Eggerthellaceae | Slackia | 0.15 | 0.178 | 55.6 | 0.04 | 0.06 | 0.51 | 0.66 |
| Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | 8.34 | 9.421 | 100 | 0.45 | 1.3 | 25.74 | 32.25 |
| Bacteroidetes | Bacteroidia | Bacteroidales | Prevotellaceae | Prevotella 9 | 23.17 | 17.55 | 80.5 | 0.11 | 1.02 | 46.51 | 56.69 |
| Bacteroidetes | Bacteroidia | Bacteroidales | Tannerellaceae | Parabacteroides | 0.51 | 1.038 | 80.5 | 0.08 | 0.14 | 1.57 | 3.71 |
| Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Clostridium sensu stricto 1 | 1.2 | 3.237 | 84 | 0.06 | 0.15 | 6.39 | 11.39 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Ruminococcus gauvreauii | 0.37 | 0.503 | 90.5 | 0.07 | 0.13 | 1.05 | 1.67 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Ruminococcus gnavus | 0.51 | 1.478 | 97 | 0.06 | 0.11 | 2.4 | 5.1 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Ruminococcus torques | 0.6 | 0.818 | 91.1 | 0.08 | 0.16 | 2.12 | 2.8 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Blautia | 5.44 | 5.65 | 100 | 1 | 1.84 | 13.88 | 20.96 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Lachnoclostridium | 1.13 | 1.103 | 98.2 | 0.2 | 0.4 | 3 | 4.28 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Lachnospiraceae NK4A136 | 0.28 | 0.43 | 86.4 | 0.05 | 0.11 | 0.83 | 1.68 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Unclassified Lachnospiraceae | 0.92 | 1.165 | 96.4 | 0.13 | 0.28 | 2.63 | 3.72 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Lachnospiraceae UCG-009 | 0.18 | 0.223 | 72.2 | 0.05 | 0.07 | 0.57 | 0.94 |
| Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | Lachnospiraceae uncultured | 0.8 | 0.888 | 93.5 | 0.11 | 0.21 | 2.04 | 3.22 |
| Firmicutes | Clostridia | Clostridiales | Peptococcaceae | Peptococcus | 0.57 | 1.37 | 65.1 | 0.09 | 0.13 | 2.64 | 4.62 |
| Firmicutes | Clostridia | Clostridiales | Peptostreptococcaceae | Peptoclostridium | 4.52 | 5.047 | 89.3 | 0.55 | 1.19 | 12.82 | 19.28 |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Butyricicoccus | 0.17 | 0.28 | 68 | 0.04 | 0.07 | 0.45 | 0.92 |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Faecalibacterium | 1.38 | 1.95 | 79.9 | 0.08 | 0.24 | 4.72 | 7.49 |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Negativibacillus | 1.19 | 1.529 | 95.3 | 0.11 | 0.23 | 3.58 | 5.69 |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Oscillibacter | 0.25 | 0.327 | 79.9 | 0.04 | 0.08 | 0.61 | 0.99 |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Ruminiclostridium 9 | 0.43 | 0.458 | 84.6 | 0.05 | 0.08 | 1.15 | 1.65 |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Ruminococcaceae UCG-014 | 0.61 | 3.432 | 74.6 | 0.06 | 0.12 | 3.07 | 9.41 |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Ruminococcaceae uncultured | 0.15 | 0.255 | 60.4 | 0.03 | 0.05 | 0.48 | 0.95 |
| Firmicutes | Erysipelotrichia | Erysipelotrichales | Erysipelotrichaceae | Catenibacterium | 2.01 | 2.179 | 56.2 | 0.16 | 0.39 | 5.6 | 7.42 |
| Firmicutes | Erysipelotrichia | Erysipelotrichales | Erysipelotrichaceae | Holdemanella | 0.85 | 2.371 | 66.9 | 0.08 | 0.17 | 3.32 | 10.01 |
| Firmicutes | Negativicutes | Selenomonadales | Veillonellaceae | Megamonas | 1.81 | 5.355 | 75.7 | 0.1 | 0.15 | 8.37 | 19.25 |
| Firmicutes | Negativicutes | Selenomonadales | Veillonellaceae | Megasphaera | 5.21 | 7.796 | 58 | 0.15 | 0.4 | 16.67 | 28.39 |
| Fusobacteria | Fusobacteriia | Fusobacteriales | Fusobacteriaceae | Fusobacterium | 3.95 | 6.149 | 88.8 | 0.11 | 0.28 | 13.43 | 21.86 |
| Proteo bacteria | Gamma proteobacteria | Betaproteobacteriales | Burkholderiaceae | Sutterella | 2.23 | 2.846 | 97 | 0.14 | 0.46 | 6.28 | 9.85 |
| Core Microbiome Total | 82.59 | 6.775 | NA | 67.65 | 74.07 | 91.59 | 93.99 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganz, H.H.; Jospin, G.; Rojas, C.A.; Martin, A.L.; Dahlhausen, K.; Kingsbury, D.D.; Osborne, C.X.; Entrolezo, Z.; Redner, S.; Ramirez, B.; et al. The Kitty Microbiome Project: Defining the Healthy Fecal “Core Microbiome” in Pet Domestic Cats. Vet. Sci. 2022, 9, 635. https://doi.org/10.3390/vetsci9110635
Ganz HH, Jospin G, Rojas CA, Martin AL, Dahlhausen K, Kingsbury DD, Osborne CX, Entrolezo Z, Redner S, Ramirez B, et al. The Kitty Microbiome Project: Defining the Healthy Fecal “Core Microbiome” in Pet Domestic Cats. Veterinary Sciences. 2022; 9(11):635. https://doi.org/10.3390/vetsci9110635
Chicago/Turabian StyleGanz, Holly H., Guillaume Jospin, Connie A. Rojas, Alex L. Martin, Katherine Dahlhausen, Dawn D. Kingsbury, Carlton X. Osborne, Zhandra Entrolezo, Syd Redner, Bryan Ramirez, and et al. 2022. "The Kitty Microbiome Project: Defining the Healthy Fecal “Core Microbiome” in Pet Domestic Cats" Veterinary Sciences 9, no. 11: 635. https://doi.org/10.3390/vetsci9110635