A Stab in the Dark: A Case Report of an Atypical Presentation of Giant Cell Arteritis (GCA)


Abstract
:1. Introduction
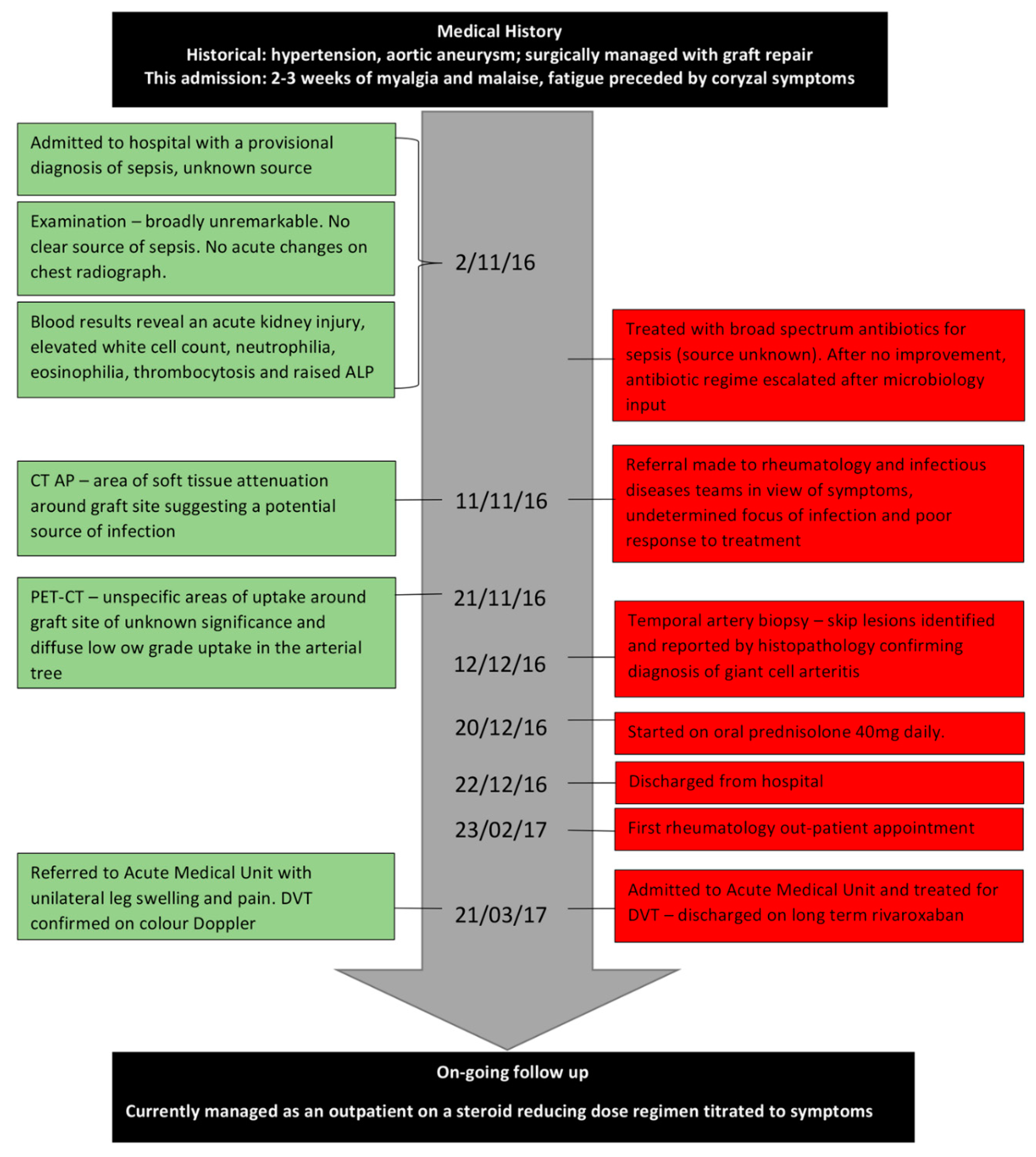
2. Case Presentation
3. Review
4. Clinical Features
5. Investigations
6. Current Management
7. Discussion
8. Conclusions
9. Patient’s Perspective
“The near daily explanations of my various symptoms by you and your team always proved to be a little mysterious and I have therefore been pleased and grateful to trust your knowledge and integrity and not to worry myself unduly regarding my lack of a physician’s skill. For that reason I am indebted to the entire team for devoting so much of their time and energy into identifying my health problem and to its continuing cure…my stay in hospital remains a blur to me but I shall always be thankful to all who did so much to make me comfortable”.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Approval
Consent to Participate
Consent to Publish
Availability of Data and Materials
Abbreviations
| GCA | Giant cell arteritis |
| TAB | Temporal artery biopsy |
| MDT | Multidisciplinary team |
| ESR | Erythrocyte sedimentation rate |
| VZV | Varicella zoster |
| CRP | c-reactive protein |
| HLA-DRB | Human leucocyte antigen, nucleotide sequence DRB |
| FDG-PET/CT | fluorodeoxyglucose positron emission tomography |
| IL- | Interleukin |
| FUO | Fever of unknown origin |
| Th- | T-helper cell |
| ALP | Alkaline phosphatase |
| IFN-γ | Interferon gamma |
| CK | Creatine Kinase |
| MRI | Magnetic resonance imaging |
| CT | Computerised tomography |
| BSR | British Society for Rheumatology |
| ANCA | Anti-neutrophil cytoplasm antibodies |
| NICE | National Institutes for Health and Care Excellence |
| TNF | Tumour necrosis factor |
| DVT | Deep vein thrombosis |
| TABUL | The Role of Ultrasound Compared to Biopsy of Temporal Arteries in the Diagnosis and Treatment of Giant Cell Arteritis: a diagnostic accuracy and cost-effectiveness study |
References
- Gagnier, J.J.; Kienle, G.; Altman, D.G.; Moher, D.; Sox, H.; Riley, D. The CARE Guidelines: Consensus-based Clinical Case Reporting Guideline Development. Glob. Adv. Health Med. 2013, 2, 38–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Smeeth, L.; Cook, C.; Hall, A.J. Incidence of diagnosed polymyalgia rheumatica and temporal arteritis in the United Kingdom, 1990–2001. Ann. Rheum. Dis. 2006, 65, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.S. Giant Cell Arteritis. Ann. Intern. Med. 2016, 165, Itc65–Itc80. [Google Scholar] [CrossRef] [PubMed]
- Della Rossa, A.; Cioffi, E.; Elefante, E.; Ferro, F.; Parma, A.; Vagelli, R.; Talarico, R. Systemic vasculitis: An annual critical digest of the most recent literature. Clin. Exp. Rheumatol. 2014, 32, S98–S105. [Google Scholar] [PubMed]
- Duhaut, P.; Pinede, L.; Demolombe-Rague, S.; Loire, R.; Seydoux, D.; Ninet, J.; Pasquier, J. Giant cell arteritis and cardiovascular risk factors: A multicenter, prospective case-control study. Arthritis Rheum. 1998, 41, 1960–1965. [Google Scholar] [CrossRef] [Green Version]
- Larsson, K.; Mellström, D.; Nordborg, C.; Odén, A. Early menopause, low body mass index, and smoking are independent risk factors for developing giant cell arteritis. Ann. Rheum. Dis. 2006, 65, 529–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Ma, S.; Zheng, W.; Tian, X.; Zeng, X. A Retrospective Study of Chinese Patients With Giant Cell Arteritis (GCA): Clinical Features and Factors Associated with Severe Ischemic Manifestations. Medicine 2016, 95, e3213. [Google Scholar] [CrossRef] [PubMed]
- Elling, P.; Olsson, A.T.; Elling, H. Synchronous variations of the incidence of temporal arteritis and polymyalgia rheumatica in different regions of Denmark; association with epidemics of Mycoplasma pneumoniae infection. J. Rheumatol. 1996, 23, 112–119. [Google Scholar] [PubMed]
- Nagel, M.A.; White, T.; Khmeleva, N.; Rempel, A.; Boyer, P.J.; Bennett, J.L.; Haller, A.; Lear-Kaul, K.; Kandasmy, B.; Amato, M.; et al. Analysis of Varicella-Zoster Virus in Temporal Arteries Biopsy Positive and Negative for Giant Cell Arteritis. JAMA Neurol. 2015, 72, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Gilden, D.; White, T.; Khmeleva, N.; Heintzman, A.; Choe, A.; Boyer, P.J.; Grose, C.; Carpenter, J.E.; Rempel, A.; Bos, N.; et al. Prevalence and distribution of VZV in temporal arteries of patients with giant cell arteritis. Neurology 2015, 84, 1948–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Geest, K.S.; Abdulahad, W.H.; Chalan, P.; Rutgers, A.; Horst, G.; Huitema, M.G.; Roffel, M.P.; Roozendaal, C.; Kluin, P.M.; Bos, N.A.; et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum. 2014, 66, 1927–1938. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B. Concise guidance: Diagnosis and management of giant cell arteritis. Clin. Med. 2010, 10, 381–386. [Google Scholar] [CrossRef]
- Keser, G.; Aksu, K.; Direskeneli, H. Discrepancies between vascular and systemic inflammation in large vessel vasculitis: An important problem revisited. Rheumatology 2018, 57, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, R.; Lee, E.; Singh, S.; Gillett, M.; Schmidt, W.A.; Bradburn, M.; Dasgupta, B.; Diamantopoulos, A.P.; Forrester-Barker, W.; Hamilton, W.; et al. The Role of Ultrasound Compared to Biopsy of Temporal Arteries in the Diagnosis and Treatment of Giant Cell Arteritis (TABUL): A diagnostic accuracy and cost-effectiveness study. Health Technol. Assess. 2016, 20, 1–238. [Google Scholar] [CrossRef] [PubMed]
- Nesher, G.; Berkun, Y.; Mates, M.; Baras, M.; Nesher, R.; Rubinow, A.; Sonnenblick, M. Risk factors for cranial ischemic complications in giant cell arteritis. Medicine 2004, 83, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Barraclough, K.; Mallen, C.D.; Helliwell, T.; Hider, S.L.; Dasgupta, B. Diagnosis and management of giant cell arteritis. Br. J. Gen. Pract. 2012, 62, 329–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kermani, T.A.; Schmidt, J.; Crowson, C.S.; Ytterberg, S.R.; Hunder, G.G.; Matteson, E.L.; Warrington, K.J. Utility of erythrocyte sedimentation rate and C-reactive protein for the diagnosis of giant cell arteritis. Semin. Arthritis Rheum. 2012, 41, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Costenbader, K.H.; Chibnik, L.B.; Schur, P.H. Discordance between erythrocyte sedimentation rate and C-reactive protein measurements: Clinical significance. Clin. Exp. Rheum. 2007, 25, 746–749. [Google Scholar]
- Gonzalez-Gay, M.A.; Garcia-Porrua, C.; Amor-Dorado, J.C.; Llorca, J. Fever in biopsy-proven giant cell arteritis: Clinical implications in a defined population. Arthritis Rheum. 2004, 51, 652–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liozon, E.; Boutros-Toni, F.; Ly, K.; Loustaud-Ratti, V.; Soria, P.; Vidal, E. Silent, or masked, giant cell arteritis is associated with a strong inflammatory response and a benign short term course. J. Rheumatol. 2003, 30, 1272–1276. [Google Scholar] [PubMed]
- Calamia, K.T.; Hunder, G.G. Giant cell arteritis (temporal arteritis) presenting as fever of undetermined origin. Arthritis Rheum. 1981, 24, 1414–1418. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Warrington, K.J. Polymyalgia Rheumatica and Giant Cell Arteritis in Older Patients. Drugs Aging 2011, 28, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Hunder, G.G.; Bloch, D.A.; Michel, B.A.; Stevens, M.B.; Arend, W.P.; Calabrese, L.H.; Edworthy, S.M.; Fauci, A.S.; Leavitt, R.Y.; Lie, J.T.; et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum. 1990, 33, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gay, M.A.; Pina, T. Giant cell arteritis and polymyalgia rheumatica: An update. Curr. Rheumatol. Rep. 2015, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Toren, A.; Weis, E.; Patel, V.; Monteith, B.; Gilberg, S.; Jordan, D. Clinical predictors of positive temporal artery biopsy. Can. J. Ophthalmol. 2016, 51, 476–481. [Google Scholar] [CrossRef] [PubMed]
- De Boysson, H.L.M.; Liozon, E.; Boutemy, J.; Maigne, G.; Ollivier, Y.; Manrique, A.; Bienvenu, B.; Aouba, A. Giant-cell arteritis without cranial manifestations. Medicine 2016, 95, e3818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton-Key, M.R.; Gallagher, P.J. False-negative temporal artery biopsy. Am. J. Surg. Pathol. 1992, 16, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Mahr, A.; Saba, M.; Kambouchner, M.; Polivka, M.; Baudrimont, M.; Brocheriou, I.; Coste, J.; Guillevin, L. Temporal artery biopsy for diagnosing giant cell arteritis: The longer, the better? Ann. Rheum. Dis. 2006, 65, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Oh, L.J.; Wong, E.; Gill, A.J.; McCluskey, P.; Smith, J.E. Value of temporal artery biopsy length in diagnosing giant cell arteritis. ANZ J. Surg. 2018, 88, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Ray-Chaudhuri, N.; Kine, D.A.; Tijani, S.O.; Parums, D.V.; Cartlidge, N.; Strong, N.P.; Dayan, M.R. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br. J. Ophthalmol. 2002, 86, 530–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arida, A.; Kyprianou, M.; Kanakis, M.; Sfikakis, P.P. The diagnostic value of ultrasonography-derived edema of the temporal artery wall in giant cell arteritis: A second meta-analysis. BMC Musculoskelet. Disord. 2010, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Ball, E.L.; Walsh, S.R.; Tang, T.Y.; Gohil, R.; Clarke, J.M. Role of ultrasonography in the diagnosis of temporal arteritis. Br. J. Surg. 2010, 97, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Karassa, F.B.; Matsagas, M.I.; Schmidt, W.A.; Ioannidis, J.P. Meta-analysis: Test performance of ultrasonography for giant-cell arteritis. Ann. Intern. Med. 2005, 142, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Floris, A.; Ponte, C.; Schmidt, W.A.; Diamantopoulos, A.P.; Pereira, C.; Piper, J.; Luqmani, R. The use of ultrasound to assess giant cell arteritis: Review of the current evidence and practical guide for the rheumatologist. Rheumatology 2018, 57, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Bley, T.A.; Reinhard, M.; Hauenstein, C.; Markl, M.; Warnatz, K.; Hetzel, A.; Uhl, M.; Vaith, P.; Langer, M. Comparison of duplex sonography and high-resolution magnetic resonance imaging in the diagnosis of giant cell (temporal) arteritis. Arthritis Rheum. 2008, 58, 2574–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klink, T.; Geiger, J.; Both, M.; Ness, T.; Heinzelmann, S.; Reinhard, M.; Holl-Ulrich, K.; Duwendag, D.; Vaith, P.; Bley, T.A. Giant cell arteritis: Diagnostic accuracy of MR imaging of superficial cranial arteries in initial diagnosis-results from a multicenter trial. Radiology 2014, 273, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vicente, A.M.; Tello-Galan, M.J.; Amo-Salas, M.; Ros-Izquierdo, J.; Jimenez-Londono, G.A.; La Rosa Salas, B.; Pradas, G.P.; Pena-Pardo, F.J.; Soriano-Castrejón, Á. Do clinical and laboratory variables have any impact on the diagnostic performance of 18F-FDG PET/CT in patients with fever of unknown origin? Ann. Nucl. Med. 2017, 32, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Blockmans, D. PET in vasculitis. Ann. N. Y. Acad. Sci. 2011, 1228, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Soussan, M.; Nicolas, P.; Schramm, C.; Katsahian, S.; Pop, G.; Fain, O.; Mekinian, A. Management of large-vessel vasculitis with FDG-PET: A systematic literature review and meta-analysis. Medicine 2015, 94, e622. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, B.D.; Gormsen, L.C.; Hansen, I.T.; Keller, K.K.; Therkildsen, P.; Hauge, E.M. Three days of high-dose glucocorticoid treatment attenuates large-vessel 18F-FDG uptake in large-vessel giant cell arteritis but with a limited impact on diagnostic accuracy. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Dejaco, C.; Ramiro, S.; Duftner, C.; Besson, F.L.; Bley, T.A.; Blockmans, D.; Brouwer, E.; Cimmino, M.A.; Clark, E.; Dasgupta, B.; et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann. Rheum. Dis. 2018, 77, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duftner, C.; Dejaco, C.; Sepriano, A.; Falzon, L.; Schmidt, W.A.; Ramiro, S. Imaging in diagnosis, outcome prediction and monitoring of large vessel vasculitis: A systematic literature review and meta-analysis informing the EULAR recommendations. RMD Open 2018, 4, e000612. [Google Scholar] [CrossRef] [PubMed]
- Charlton, R. Optimal management of giant cell arteritis and polymyalgia rheumatica. Ther. Clin. Risk Manag. 2012, 8, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Borg, F.A.; Hassan, N.; Alexander, L.; Barraclough, K.; Bourke, B.; Fulcher, J.; Hollywood, J.; Hutchings, A.; James, P.; et al. BSR and BHPR guidelines for the management of giant cell arteritis. Rheumatology 2010, 49, 1594–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesher, G.; Berkun, Y.; Mates, M.; Baras, M.; Rubinow, A.; Sonnenblick, M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum. 2004, 50, 1332–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NICE. Giant Cell Arteritis. 2014. Available online: www.nice.org.uk/giant-cell-arteritis#!scenario (accessed on 24 June 2018).
- Fardet, L. Glucocorticoid-induced adverse events in patients with giant cell arteritis or polymyalgia rheumatica. La Revue Med. Intern. 2013, 34, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Mahr, A.D.; Jover, J.A.; Spiera, R.F.; Hernandez-Garcia, C.; Fernandez-Gutierrez, B.; Lavalley, M.P.; Merkel, P.A. Adjunctive methotrexate for treatment of giant cell arteritis: An individual patient data meta-analysis. Arthritis Rheum. 2007, 56, 2789–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watelet, B.; Samson, M.; de Boysson, H.; Bienvenu, B. Treatment of giant-cell arteritis, a literature review. Mod. Rheumatol. 2017, 27, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Villiger, P.M.; Adler, S.; Kuchen, S.; Wermelinger, F.; Dan, D.; Fiege, V.; Bütikofer, L.; Seitz, M.; Reichenbach, S. Tocilizumab for induction and maintenance of remission in giant cell arteritis: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1921–1927. [Google Scholar] [CrossRef]
- Stone, J.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Cid, M.C.; Dasgupta, B.; Rech, J.; Salvarani, C.; et al. Trial of Tocilizumab in Giant-Cell Arteritis. N. Eng. J. Med. 2017, 377, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Takeda, R.; Mizooka, M.; Kobayashi, T.; Kishikawa, N.; Yokobayashi, K.; Kanno, K.; Tazuma, S. Key diagnostic features of fever of unknown origin: Medical history and physical findings. J. Gen. Fam. Med. 2017, 18, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenone, T.; Puget, M. Dry cough is a frequent manifestation of giant cell arteritis. Rheumatol. Int. 2013, 33, 2165–2168. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.H.H.; Okada, T.; Terui, T. Hypereosinophilic syndrome with various skin lesions and juvenile temporal arteritis. Clin. Exp. Dermatol. 2009, 34, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Cavazza, A.; Muratore, F.; Boiardi, L.; Restuccia, G.; Pipitone, N.; Pazzola, G.; Tagliavini, E.; Ragazzi, M.; Rossi, G.; Salvarani, C. Inflamed temporal artery: Histologic findings in 354 biopsies, with clinical correlations. Am. J. Surg. Pathol. 2014, 38, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Ezeonyeji, A.N.; Borg, F.A.; Dasgupta, B. Delays in recognition and management of giant cell arteritis: Results from a retrospective audit. Clin. Rheumatol. 2011, 30, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Nuenninghoff, D.M.; Hunder, G.G.; Christianson, T.J.; McClelland, R.L.; Matteson, E.L. Mortality of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: A population-based study over 50 years. Arthritis Rheum. 2003, 48, 3532–3537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avina-Zubieta, J.A.; Bhole, V.M.; Amiri, N.; Sayre, E.C.; Choi, H.K. The risk of deep venous thrombosis and pulmonary embolism in giant cell arteritis: A general population-based study. Ann. Rheum. Dis. 2016, 75, 148–154. [Google Scholar] [CrossRef] [PubMed]
- De Boysson, H.; Aide, N.; Liozon, E.; Lambert, M.; Parienti, J.J.; Monteil, J.; Huglo, D.; Bienvenu, B.; Manrique, A.; Aouba, A. Repetitive (18)F-FDG-PET/CT in patients with large-vessel giant-cell arteritis and controlled disease. Eur. J. Intern. Med. 2017, 46, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, C.; Soriano, A.; Muratore, F.; Shoenfeld, Y.; Blockmans, D. Is PET/CT essential in the diagnosis and follow-up of temporal arteritis? Autoimmun. Rev. 2017, 16, 1125–1130. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Units of Measurement | Normal Range | Patient Values on Admission | Patient Values on Discharge | |
|---|---|---|---|---|
| Haemoglobin | g/L | 130–170 | 138 | 121 |
| White cell count | 109/L | 4.0–11.0 | 26.2 | 11.2 |
| Platelets | 109/L | 150–400 | 576 | 308 |
| Neutrophil | 109/L | 2.0–7.5 | 21.9 | 7.1 |
| Eosinophil | 109/L | 0.0–0.5 | 1.7 | 0.7 |
| ESR | mm/h | 1–30 | 22 | 5 |
| CRP | mg/L | 0–7.5 | 245 | 33 |
| Urea | mmol/L | 2.5–7.8 | 24.2 | 12.4 |
| Creatinine | μmol/L | 80–115 | 154 | 130 |
| Creatine Kinase (CK) | U/L | 40–320 | 83 | N/A |
| ALT | U/L | 10–40 | 30 | 11 |
| ALP | U/L | 30–130 | 185 | 88 |
| Albumin | g/L | 35–50 | 19 | 25 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCausland, B.; Desai, D.; Havard, D.; Kaur, Y.; Yener, A.; Bradley, E.; Patel, H.P. A Stab in the Dark: A Case Report of an Atypical Presentation of Giant Cell Arteritis (GCA). Geriatrics 2018, 3, 36. https://doi.org/10.3390/geriatrics3030036
McCausland B, Desai D, Havard D, Kaur Y, Yener A, Bradley E, Patel HP. A Stab in the Dark: A Case Report of an Atypical Presentation of Giant Cell Arteritis (GCA). Geriatrics. 2018; 3(3):36. https://doi.org/10.3390/geriatrics3030036
Chicago/Turabian StyleMcCausland, Beth, David Desai, David Havard, Yasmin Kaur, Asalet Yener, Emma Bradley, and Harnish P. Patel. 2018. "A Stab in the Dark: A Case Report of an Atypical Presentation of Giant Cell Arteritis (GCA)" Geriatrics 3, no. 3: 36. https://doi.org/10.3390/geriatrics3030036