
Cellular Senescence in Cardiovascular Diseases: From Pathogenesis to Therapeutic Challenges
Abstract
:1. Introduction
2. Definition and Mechanisms of CS
2.1. Replicative CS
2.2. Premature CS
2.2.1. DNA Damage-Induced Senescence
2.2.2. Oncogene-Induced Senescence
2.2.3. Oxidative Stress-Induced Senescence
2.2.4. Paracrine Senescence
2.2.5. Mitochondrial Dysfunction-Associated Senescence (MiDAS)
2.2.6. Epigenetically Induced Senescence
2.2.7. Other Factors
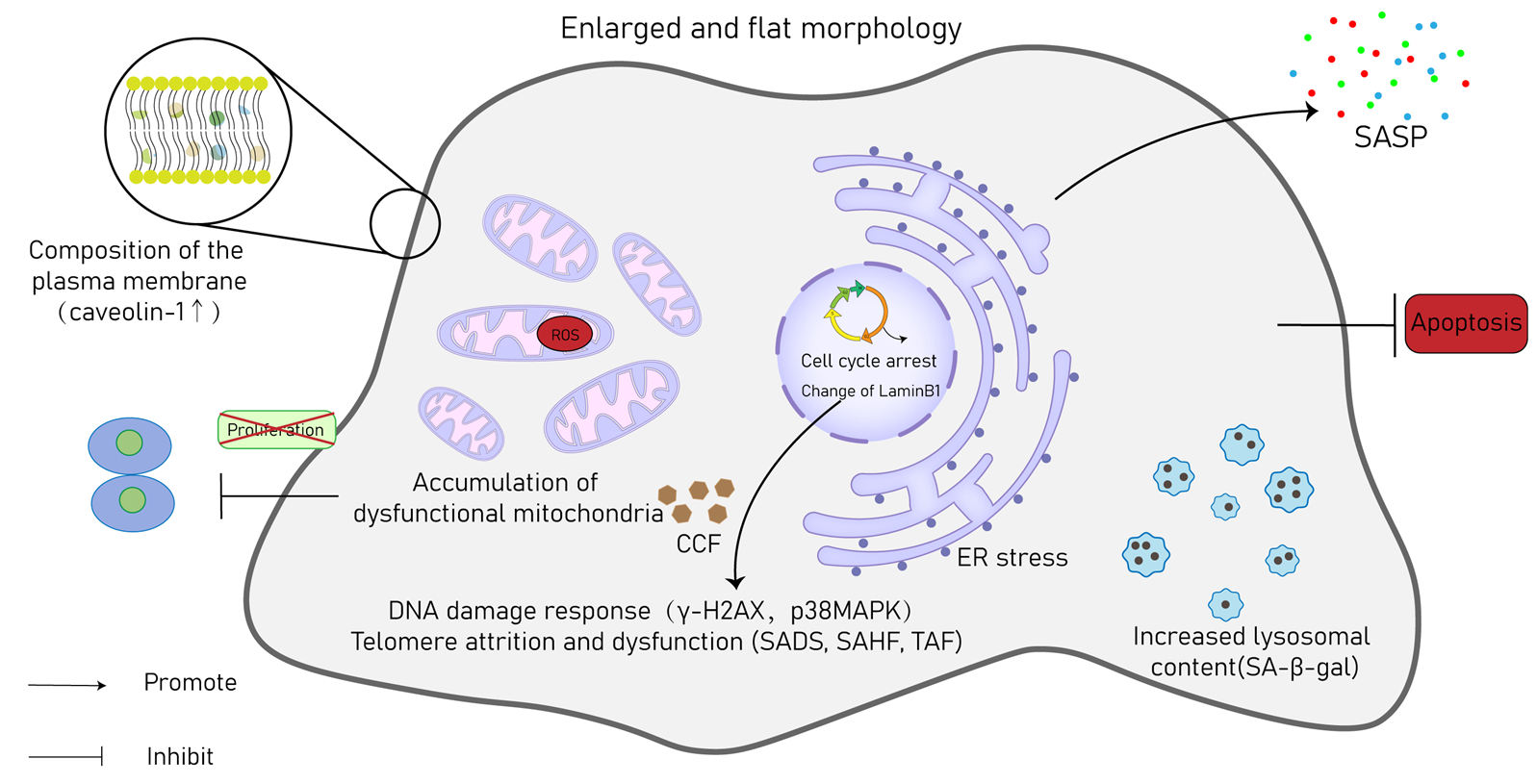
3. Hallmarks of CS
4. Senescence in Specific Cardiac Cell Types
4.1. Cardiomyocytes
4.2. Endothelial Cells
4.3. Cardiac Fibroblasts
4.4. VSMCs
4.5. Immune Cells
4.6. Cardiac Progenitor Cells
5. Physiology and Pathophysiology of CS in the Cardiovascular System
5.1. Senescence in Heart Development
5.2. Senescence in Cardiac Pathology
5.2.1. HF
5.2.2. MI
5.2.3. Hypertrophic Cardiomyopathy
5.2.4. Cancer Therapy-Induced Cardiotoxicity
5.2.5. Cardiac Fibrosis
5.2.6. Diabetic Cardiomyopathy
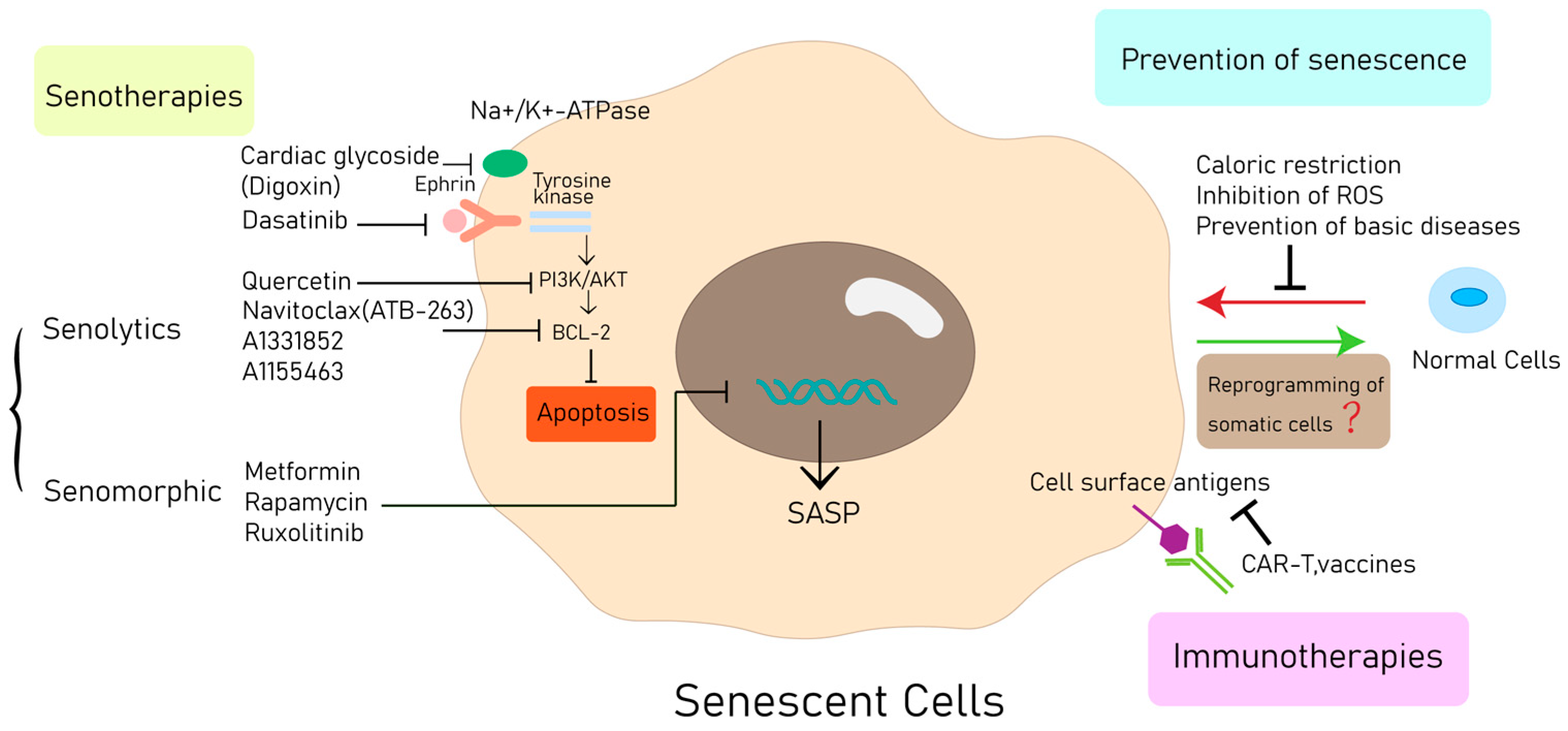
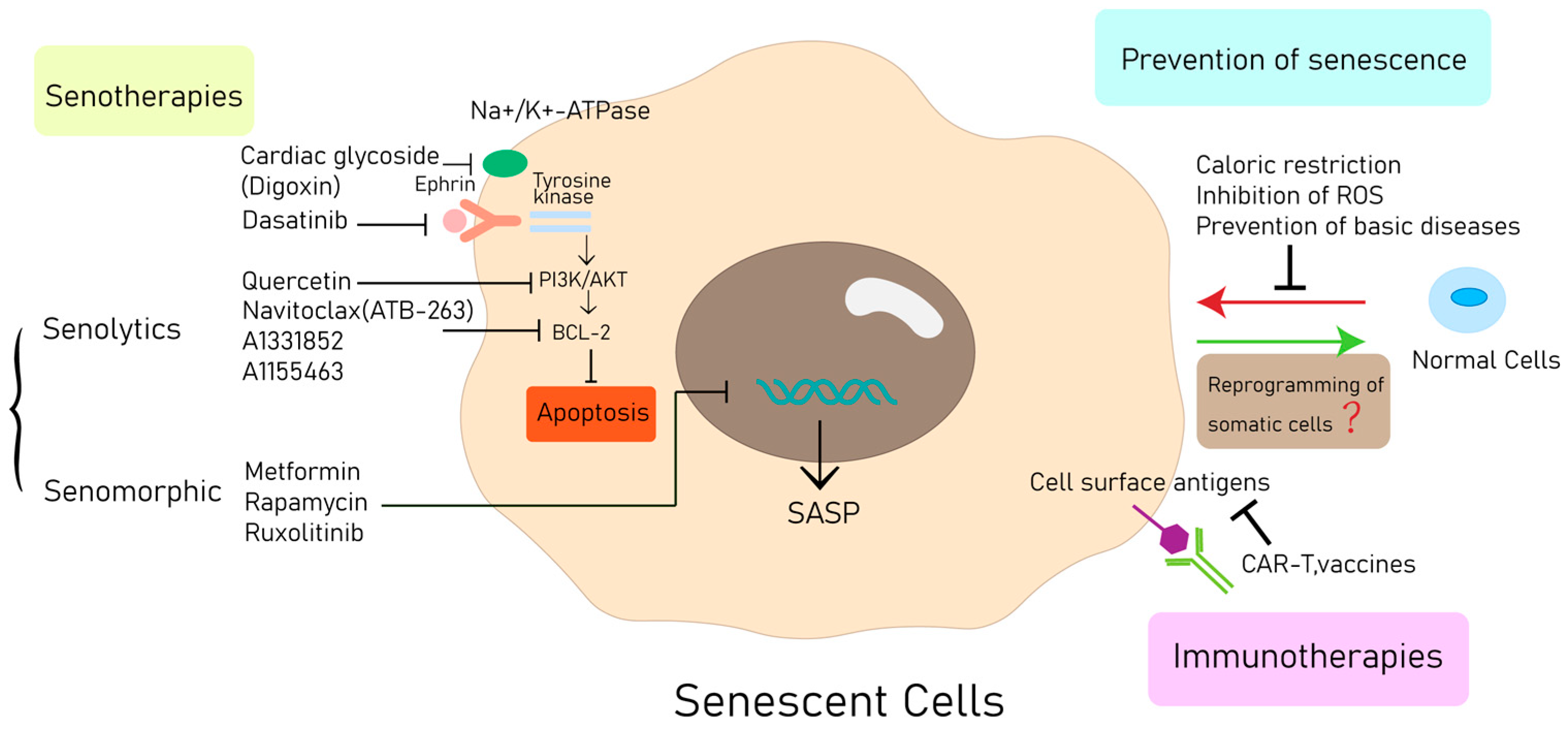
6. Therapeutic Approaches
6.1. Prevention of Senescence
6.2. Senotherapies
6.2.1. Senolytics
6.2.2. Manipulation of the SASP
6.3. Immune Activation of Target SNCs
6.4. Bypassing and Reverting Senescence
7. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning |
| CS | cellular senescence |
| SASP | senescence-associated secretory phenotype |
| CVDs | cardiovascular diseases |
| SIPS | stress-induced premature senescence |
| RS | replicative senescence |
| DDR | DNA damage response |
| hTERT | human telomerase reverse transcriptase |
| SSBs | single-strand breaks |
| DSBs | double-strand breaks |
| ATM | ataxia–telangiectasia mutated |
| ATR | ataxia–telangiectasia Rad3-related |
| γH2AX | phosphorylated histone H2AX |
| CDKN1A | CDK inhibitor p21 |
| cGAS | cyclic GMP–AMP synthase |
| STING | stimulator of interferon genes |
| TAFs | telomere-associated foci |
| mTOR | mammalian target of rapamycin |
| ROS | reactive oxygen species |
| OIS | oncogene-induced senescence |
| mtDNA | mitochondrial DNA |
| ECM | extracellular membrane |
| mi-RNA | microRNAs |
| lncRNA | long non-coding RNAs |
| EndoG | endonuclease G |
| SA-β-gal | senescence-associated β-galactosidase |
| SBB | Sudan Black B |
| CCFs | cytoplasmic chromatin fragments |
| p38MAPK | p38 mitogen-activated protein kinase |
| SAHF | senescence-associated heterochromatin foci |
| SADs | senescence-associated distention of satellites |
| H2O2 | hydrogen peroxide |
| BrdU | bromodeoxyuridine |
| EdU | ethynyl deoxyuridine |
| VSMCs | vascular smooth muscle cells |
| DMD | Duchenne muscular dystrophy |
| LMNA | lamin A |
| ET-1 | endothelin-1 |
| HF | heart failure |
| HFpEF | heart failure with preserved ejection fraction |
| MMPs | matrix metalloproteinases |
| MI | myocardial infarction |
| CAD | coronary artery disease |
| T1DM | type 1 diabetes |
| STZ | streptozotocin |
| HIF-1α | hypoxia-inducible factor-1α |
| SNCs | senescent cells |
| AMPK | AMP-activated protein kinase |
| SCAPs | senescent cell anti-apoptotic pathways |
| BCL-2 | B-cell lymphoma 2 |
| NF-κB | nuclear factor kappa B |
| HSP90 | heat shock protein 90 |
| FDA | Food and Drug Administration |
| TAME | Targeting Aging with Metformin |
| AFAR | American Federation for Aging Research |
| CAR-T | chimeric antigen receptor T cell |
| CTL | cytotoxic T lymphocyte |
| NK | natural killer |
| DC | dendritic cell |
| GPNMB | glycoprotein non-metastatic melanoma protein B |
| ADCs | antibody–drug conjugates |
| TGF-β | transforming growth factor-β |
| SMAD | small mother against decapentaplegic |
| AMPK | adenosine 5‘-monophosphate (AMP)-activated protein kinase |
| Rb | retinoblastoma protein |
| MiDAS | mitochondrial dysfunction-associated senescence |
| IGF-1 | insulin-like growth factor 1 |
| CPCS | cardiac progenitor cells |
References
- Mehdizadeh, M.; Aguilar, M.; Thorin, E.; Ferbeyre, G.; Nattel, S. The role of cellular senescence in cardiac disease: Basic biology and clinical relevance. Nat. Rev. Cardiol. 2022, 19, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Vasileiou, P.V.S.; Papaspyropoulos, A.; Hazapis, O.; Petty, R.; Demaria, M.; Gorgoulis, V.G. Cellular senescence and cardiovascular diseases: Moving to the “heart” of the problem. Physiol. Rev. 2023, 103, 609–647. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Laredo, M.; Waldmann, V.; Khairy, P.; Nattel, S. Age as a Critical Determinant of Atrial Fibrillation: A Two-sided Relationship. Can. J. Cardiol. 2018, 34, 1396–1406. [Google Scholar] [CrossRef]
- Kuo, C.L.; Pilling, L.C.; Kuchel, G.A.; Ferrucci, L.; Melzer, D. Telomere length and aging-related outcomes in humans: A Mendelian randomization study in 261,000 older participants. Aging Cell 2019, 18, e13017. [Google Scholar] [CrossRef]
- Noly, P.E.; Labbé, P.; Thorin-Trescases, N.; Fortier, A.; Nguyen, A.; Thorin, E.; Carrier, M. Reduction of plasma angiopoietin-like 2 after cardiac surgery is related to tissue inflammation and senescence status of patients. J. Thorac. Cardiovasc. Surg. 2019, 158, 792–802.e5. [Google Scholar] [CrossRef]
- Gude, N.A.; Broughton, K.M.; Firouzi, F.; Sussman, M.A. Cardiac ageing: Extrinsic and intrinsic factors in cellular renewal and senescence. Nat. Rev. Cardiol. 2018, 15, 523–542. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hu, C.; Zhang, X.; Teng, T.; Ma, Z.G.; Tang, Q.Z. Cellular Senescence in Cardiovascular Diseases: A Systematic Review. Aging Dis. 2022, 13, 103–128. [Google Scholar] [CrossRef]
- Serrano, M.; Blasco, M.A. Putting the stress on senescence. Curr. Opin. Cell Biol. 2001, 13, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Blackburn, E.H. Structure and function of telomeres. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Gorospe, M. Noncoding RNAs Controlling Telomere Homeostasis in Senescence and Aging. Trends Mol. Med. 2020, 26, 422–433. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef]
- Masutomi, K.; Yu, E.Y.; Khurts, S.; Ben-Porath, I.; Currier, J.L.; Metz, G.B.; Brooks, M.W.; Kaneko, S.; Murakami, S.; DeCaprio, J.A.; et al. Telomerase maintains telomere structure in normal human cells. Cell 2003, 114, 241–253. [Google Scholar] [CrossRef]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.S. Genomic damage and its repair in young and aging brain. Mol. Neurobiol. 1993, 7, 23–48. [Google Scholar] [CrossRef]
- Soto-Palma, C.; Niedernhofer, L.J.; Faulk, C.D.; Dong, X. Epigenetics, DNA damage, and aging. J. Clin. Investig. 2022, 132, e158446. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Firsanov, D.; Zhang, Z.; Cheng, Y.; Luo, L.; Tombline, G.; Tan, R.; Simon, M.; Henderson, S.; Steffan, J.; et al. SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 2019, 177, 622–638.e22. [Google Scholar] [CrossRef] [PubMed]
- Shmulevich, R.; Krizhanovsky, V. Cell Senescence, DNA Damage, and Metabolism. Antioxid. Redox Signal. 2021, 34, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; et al. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Ding, J.; Meng, L.H. Oncogene-induced senescence: A double edged sword in cancer. Acta Pharmacol. Sin. 2018, 39, 1553–1558. [Google Scholar] [CrossRef]
- Wei, S.; Wei, S.; Sedivy, J.M. Expression of catalytically active telomerase does not prevent premature senescence caused by overexpression of oncogenic Ha-Ras in normal human fibroblasts. Cancer Res. 1999, 59, 1539–1543. [Google Scholar]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes. Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Izpisúa Belmonte, J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef]
- Marión, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Lowe, S.W. Stem cells: The promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Ma, D.; Stokes, K.; Mahngar, K.; Domazet-Damjanov, D.; Sikorska, M.; Pandey, S. Inhibition of stress induced premature senescence in presenilin-1 mutated cells with water soluble Coenzyme Q10. Mitochondrion 2014, 17, 106–115. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Dorn, G.W., 2nd. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 2011, 109, 1327–1331. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef]
- Floyd, R.A.; West, M.; Hensley, K. Oxidative biochemical markers; clues to understanding aging in long-lived species. Exp. Gerontol. 2001, 36, 619–640. [Google Scholar] [CrossRef]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; Vanderkraats, N.D.; Shah, P.P.; van Tuyn, J.; Singh Rai, T.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef]
- Lyu, G.; Guan, Y.; Zhang, C.; Zong, L.; Sun, L.; Huang, X.; Huang, L.; Zhang, L.; Tian, X.L.; Zhou, Z.; et al. TGF-β signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat. Commun. 2018, 9, 2560. [Google Scholar] [CrossRef]
- Florio, M.C.; Magenta, A.; Beji, S.; Lakatta, E.G.; Capogrossi, M.C. Aging, MicroRNAs, and Heart Failure. Curr. Probl. Cardiol. 2020, 45, 100406. [Google Scholar] [CrossRef]
- Lozano-Vidal, N.; Bink, D.I.; Boon, R.A. Long noncoding RNA in cardiac aging and disease. J. Mol. Cell Biol. 2019, 11, 860–867. [Google Scholar] [CrossRef]
- Rota, M.; Goichberg, P.; Anversa, P.; Leri, A. Aging Effects on Cardiac Progenitor Cell Physiology. Compr. Physiol. 2015, 5, 1775–1814. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Yamagishi, T.; Nakamura, T.; Ohyama, Y.; Aizawa, H.; Suga, T.; Matsumura, Y.; Masuda, H.; Kurabayashi, M.; Kuro-o, M.; et al. Klotho protein protects against endothelial dysfunction. Biochem. Biophys. Res. Commun. 1998, 248, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [PubMed]
- Ball, A.J.; Levine, F. Telomere-independent cellular senescence in human fetal cardiomyocytes. Aging Cell 2005, 4, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Bai, L.; Liu, L.; Fu, J.; Wu, K.; Liu, H.; Liu, Y.; Qi, B.; Qi, B. Redd1 knockdown prevents doxorubicin-induced cardiac senescence. Aging 2021, 13, 13788–13806. [Google Scholar] [CrossRef]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Minamino, T.; Yoshida, T.; Tateno, K.; Miyauchi, H.; Zou, Y.; Toko, H.; Komuro, I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation 2003, 108, 2264–2269. [Google Scholar] [CrossRef]
- Ohno-Iwashita, Y.; Shimada, Y.; Hayashi, M.; Inomata, M. Plasma membrane microdomains in aging and disease. Geriatr. Gerontol. Int. 2010, 10 (Suppl. S1), S41–S52. [Google Scholar] [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? eBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef]
- Kopp, H.G.; Hooper, A.T.; Shmelkov, S.V.; Rafii, S. Beta-galactosidase staining on bone marrow. The osteoclast pitfall. Histol. Histopathol. 2007, 22, 971–976. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef]
- Takahashi, A.; Imai, Y.; Yamakoshi, K.; Kuninaka, S.; Ohtani, N.; Yoshimoto, S.; Hori, S.; Tachibana, M.; Anderton, E.; Takeuchi, T.; et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C(Cdh1) in senescent cells. Mol. Cell 2012, 45, 123–131. [Google Scholar] [CrossRef]
- Anderson, R.; Richardson, G.D.; Passos, J.F. Mechanisms driving the ageing heart. Exp. Gerontol. 2018, 109, 5–15. [Google Scholar] [CrossRef]
- Swanson, E.C.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013, 203, 929–942. [Google Scholar] [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Lee, R.T.; Garbern, J.C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 2022, 118, 1173–1187. [Google Scholar] [CrossRef]
- Colliva, A.; Braga, L.; Giacca, M.; Zacchigna, S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J. Physiol. 2020, 598, 2923–2939. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.U.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef]
- Lee, O.H.; Woo, Y.M.; Moon, S.; Lee, J.; Park, H.; Jang, H.; Park, Y.Y.; Bae, S.K.; Park, K.H.; Heo, J.H.; et al. Sirtuin 6 deficiency induces endothelial cell senescence via downregulation of forkhead box M1 expression. Aging 2020, 12, 20946–20967. [Google Scholar] [CrossRef]
- Saucerman, J.J.; Tan, P.M.; Buchholz, K.S.; McCulloch, A.D.; Omens, J.H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019, 16, 361–378. [Google Scholar] [CrossRef]
- Civitarese, R.A.; Kapus, A.; McCulloch, C.A.; Connelly, K.A. Role of integrins in mediating cardiac fibroblast-cardiomyocyte cross talk: A dynamic relationship in cardiac biology and pathophysiology. Basic Res. Cardiol. 2017, 112, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.H.; Du, J. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef]
- Daseke, M.J., 2nd; Tenkorang, M.A.A.; Chalise, U.; Konfrst, S.R.; Lindsey, M.L. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol. 2020, 91–92, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Donastorg Sosa, L.; Gill, E.; et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 2020, 19, e13249. [Google Scholar] [CrossRef]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; De Meyer, G.R.; Martinet, W.; Schrijvers, D.M. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: Effects of telomerase and oxidative stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef]
- Saker, M.; Lipskaia, L.; Marcos, E.; Abid, S.; Parpaleix, A.; Houssaini, A.; Validire, P.; Girard, P.; Noureddine, H.; Boyer, L.; et al. Osteopontin, a Key Mediator Expressed by Senescent Pulmonary Vascular Cells in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Farb, A.; Malcom, G.T.; Liang, Y.H.; Smialek, J.; Virmani, R. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N. Engl. J. Med. 1997, 336, 1276–1282. [Google Scholar] [CrossRef]
- Libby, P.; Nahrendorf, M.; Pittet, M.J.; Swirski, F.K. Diversity of denizens of the atherosclerotic plaque: Not all monocytes are created equal. Circulation 2008, 117, 3168–3170. [Google Scholar] [CrossRef] [PubMed]
- Newby, A.C.; George, S.J.; Ismail, Y.; Johnson, J.L.; Sala-Newby, G.B.; Thomas, A.C. Vulnerable atherosclerotic plaque metalloproteinases and foam cell phenotypes. Thromb. Haemost. 2009, 101, 1006–1011. [Google Scholar]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Oguchi, A.; Fukushima, Y.; Masuda, K.; Toriu, N.; Taniguchi, K.; Yoshikawa, T.; Cui, X.; Kondo, M.; Hosoi, T.; et al. CD153/CD30 signaling promotes age-dependent tertiary lymphoid tissue expansion and kidney injury. J. Clin. Investig. 2022, 132, e146071. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Brouilette, S.W.; Whittaker, A.; Stevens, S.E.; van der Harst, P.; Goodall, A.H.; Samani, N.J. Telomere length is shorter in healthy offspring of subjects with coronary artery disease: Support for the telomere hypothesis. Heart 2008, 94, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.C.; Yu, H.T.; Lim, B.J.; Koh, M.J.; Lee, J.; Chang, D.Y.; Choi, Y.S.; Lee, S.H.; Kang, S.M.; Jang, Y.; et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 2013, 62, 126–133. [Google Scholar] [CrossRef]
- Delgobo, M.; Heinrichs, M.; Hapke, N.; Ashour, D.; Appel, M.; Srivastava, M.; Heckel, T.; Spyridopoulos, I.; Hofmann, U.; Frantz, S.; et al. Terminally Differentiated CD4(+) T Cells Promote Myocardial Inflammaging. Front. Immunol. 2021, 12, 584538. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, N.K.; Lim, H.J.; Ji, S.T.; Kim, Y.J.; Jang, W.B.; Kim, D.Y.; Kang, S.; Yun, J.; Ha, J.S.; et al. Pharmacological inhibition of mTOR attenuates replicative cell senescence and improves cellular function via regulating the STAT3-PIM1 axis in human cardiac progenitor cells. Exp. Mol. Med. 2020, 52, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Cesselli, D.; Beltrami, A.P.; D’Aurizio, F.; Marcon, P.; Bergamin, N.; Toffoletto, B.; Pandolfi, M.; Puppato, E.; Marino, L.; Signore, S.; et al. Effects of age and heart failure on human cardiac stem cell function. Am. J. Pathol. 2011, 179, 349–366. [Google Scholar] [CrossRef]
- Suda, M.; Paul, K.H.; Minamino, T.; Miller, J.D.; Lerman, A.; Ellison-Hughes, G.M.; Tchkonia, T.; Kirkland, J.L. Senescent Cells: A Therapeutic Target in Cardiovascular Diseases. Cells 2023, 12, 1296. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.H.; Chen, H.Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front. Endocrinol. 2020, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Lorda-Diez, C.I.; Solis-Mancilla, M.E.; Sanchez-Fernandez, C.; Garcia-Porrero, J.A.; Hurle, J.M.; Montero, J.A. Cell senescence, apoptosis and DNA damage cooperate in the remodeling processes accounting for heart morphogenesis. J. Anat. 2019, 234, 815–829. [Google Scholar] [CrossRef]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, X.; Huang, X.; Xia, Q.; Chen, Z.; Zhang, X.; Yang, D.; Geng, Y.J. Pax8 plays a pivotal role in regulation of cardiomyocyte growth and senescence. J. Cell. Mol. Med. 2016, 20, 644–654. [Google Scholar] [CrossRef]
- Roy, A.L.; Sierra, F.; Howcroft, K.; Singer, D.S.; Sharpless, N.; Hodes, R.J.; Wilder, E.L.; Anderson, J.M. A Blueprint for Characterizing Senescence. Cell 2020, 183, 1143–1146. [Google Scholar] [CrossRef]
- Jalloh, M.B.; Averbuch, T.; Kulkarni, P.; Granger, C.B.; Januzzi, J.L.; Zannad, F.; Yeh, R.W.; Yancy, C.W.; Fonarow, G.C.; Breathett, K.; et al. Bridging Treatment Implementation Gaps in Patients with Heart Failure: JACC Focus Seminar 2/3. J. Am. Coll. Cardiol. 2023, 82, 544–558. [Google Scholar] [CrossRef]
- Kosugi, R.; Shioi, T.; Watanabe-Maeda, K.; Yoshida, Y.; Takahashi, K.; Machida, Y.; Izumi, T. Angiotensin II receptor antagonist attenuates expression of aging markers in diabetic mouse heart. Circ. J. 2006, 70, 482–488. [Google Scholar] [CrossRef]
- Din, S.; Konstandin, M.H.; Johnson, B.; Emathinger, J.; Völkers, M.; Toko, H.; Collins, B.; Ormachea, L.; Samse, K.; Kubli, D.A.; et al. Metabolic dysfunction consistent with premature aging results from deletion of Pim kinases. Circ. Res. 2014, 115, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. Biomarkers in heart failure. N. Engl. J. Med. 2008, 358, 2148–2159. [Google Scholar] [CrossRef]
- Inuzuka, Y.; Okuda, J.; Kawashima, T.; Kato, T.; Niizuma, S.; Tamaki, Y.; Iwanaga, Y.; Yoshida, Y.; Kosugi, R.; Watanabe-Maeda, K.; et al. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation 2009, 120, 1695–1703. [Google Scholar] [CrossRef]
- Cui, S.; Xue, L.; Yang, F.; Dai, S.; Han, Z.; Liu, K.; Liu, B.; Yuan, Q.; Cui, Z.; Zhang, Y.; et al. Postinfarction Hearts Are Protected by Premature Senescent Cardiomyocytes Via GATA 4-Dependent CCN 1 Secretion. J. Am. Heart Assoc. 2018, 7, e009111. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef]
- Morin, D.; Long, R.; Panel, M.; Laure, L.; Taranu, A.; Gueguen, C.; Pons, S.; Leoni, V.; Caccia, C.; Vatner, S.F.; et al. Hsp22 overexpression induces myocardial hypertrophy, senescence and reduced life span through enhanced oxidative stress. Free Radic. Biol. Med. 2019, 137, 194–200. [Google Scholar] [CrossRef]
- Li, Y.; Ma, Y.; Song, L.; Yu, L.; Zhang, L.; Zhang, Y.; Xing, Y.; Yin, Y.; Ma, H. SIRT3 deficiency exacerbates p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int. J. Mol. Med. 2018, 41, 3517–3526. [Google Scholar] [CrossRef]
- Zha, Z.; Wang, J.; Wang, X.; Lu, M.; Guo, Y. Involvement of PINK1/Parkin-mediated mitophagy in AGE-induced cardiomyocyte aging. Int. J. Cardiol. 2017, 227, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef]
- Nattel, S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin. Electrophysiol. 2017, 3, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Sawaki, D.; Czibik, G.; Pini, M.; Ternacle, J.; Suffee, N.; Mercedes, R.; Marcelin, G.; Surenaud, M.; Marcos, E.; Gual, P.; et al. Visceral Adipose Tissue Drives Cardiac Aging Through Modulation of Fibroblast Senescence by Osteopontin Production. Circulation 2018, 138, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Zhang, Z.; Zheng, C.; Wintergerst, K.A.; Keller, B.B.; Cai, L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: Preclinical and clinical evidence. Nat. Rev. Cardiol. 2020, 17, 585–607. [Google Scholar] [CrossRef]
- Gu, J.; Wang, S.; Guo, H.; Tan, Y.; Liang, Y.; Feng, A.; Liu, Q.; Damodaran, C.; Zhang, Z.; Keller, B.B.; et al. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis. 2018, 9, 82. [Google Scholar] [CrossRef]
- Childs, B.G.; Li, H.; van Deursen, J.M. Senescent cells: A therapeutic target for cardiovascular disease. J. Clin. Investig. 2018, 128, 1217–1228. [Google Scholar] [CrossRef]
- Song, P.; Zhao, Q.; Zou, M.H. Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res. Rev. 2020, 60, 101072. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef]
- Macip, S.; Igarashi, M.; Fang, L.; Chen, A.; Pan, Z.Q.; Lee, S.W.; Aaronson, S.A. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef]
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Suda, M.; Wakasugi, T.; Nakao, M.; Minamino, T. Catecholamine-Induced Senescence of Endothelial Cells and Bone Marrow Cells Promotes Cardiac Dysfunction in Mice. Int. Heart J. 2018, 59, 837–844. [Google Scholar] [CrossRef]
- Sung, J.Y.; Kim, S.G.; Kim, J.R.; Choi, H.C. Prednisolone suppresses adriamycin-induced vascular smooth muscle cell senescence and inflammatory response via the SIRT1-AMPK signaling pathway. PLoS ONE 2020, 15, e0239976. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Chen, H.; Xie, C.; Hou, M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging 2020, 12, 8241–8260. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Acelajado, M.C.; Pisoni, R.; Dudenbostel, T.; Dell’Italia, L.J.; Cartmill, F.; Zhang, B.; Cofield, S.S.; Oparil, S.; Calhoun, D.A. Refractory hypertension: Definition, prevalence, and patient characteristics. J. Clin. Hypertens. 2012, 14, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Kajstura, J.; Torella, D.; Urbanek, K.; Heleniak, H.; Colussi, C.; Di Meglio, F.; Nadal-Ginard, B.; Frustaci, A.; Leri, A.; et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ. Res. 2003, 93, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Kosinsky, R.L.; Atkinson, E.J.; Doolittle, M.L.; Zhang, X.; LeBrasseur, N.K.; Pignolo, R.J.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat. Commun. 2022, 13, 4827. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Jiang, Y.H.; Jiang, L.Y.; Wang, Y.C.; Ma, D.F.; Li, X. Quercetin Attenuates Atherosclerosis via Modulating Oxidized LDL-Induced Endothelial Cellular Senescence. Front. Pharmacol. 2020, 11, 512. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. eBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Xu, Q.; Fu, Q.; Li, Z.; Liu, H.; Wang, Y.; Lin, X.; He, R.; Zhang, X.; Ju, Z.; Campisi, J.; et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat. Metab. 2021, 3, 1706–1726. [Google Scholar] [CrossRef] [PubMed]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular senescence and senolytics: The path to the clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Huffman, D.M.; Justice, J.N.; Stout, M.B.; Kirkland, J.L.; Barzilai, N.; Austad, S.N. Evaluating Health Span in Preclinical Models of Aging and Disease: Guidelines, Challenges, and Opportunities for Geroscience. J. Gerontol. Biol. Sci. Med. Sci. 2016, 71, 1395–1406. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Wang, G.; Zhu, X.; Xie, W.; Han, P.; Li, K.; Sun, Z.; Wang, Y.; Chen, C.; Song, R.; Cao, C.; et al. Rad as a novel regulator of excitation-contraction coupling and beta-adrenergic signaling in heart. Circ. Res. 2010, 106, 317–327. [Google Scholar] [CrossRef]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 2015, 4, e12997. [Google Scholar] [CrossRef] [PubMed]
- Suda, M.; Shimizu, I.; Katsuumi, G.; Hsiao, C.L.; Yoshida, Y.; Matsumoto, N.; Yoshida, Y.; Katayama, A.; Wada, J.; Seki, M.; et al. Glycoprotein nonmetastatic melanoma protein B regulates lysosomal integrity and lifespan of senescent cells. Sci. Rep. 2022, 12, 6522. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Nakagami, H.; Hayashi, H.; Ikeda, Y.; Sun, J.; Tenma, A.; Tomioka, H.; Kawano, T.; Shimamura, M.; Morishita, R.; et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 2020, 11, 2482. [Google Scholar] [CrossRef]
- Poblocka, M.; Bassey, A.L.; Smith, V.M.; Falcicchio, M.; Manso, A.S.; Althubiti, M.; Sheng, X.; Kyle, A.; Barber, R.; Frigerio, M.; et al. Targeted clearance of senescent cells using an antibody-drug conjugate against a specific membrane marker. Sci. Rep. 2021, 11, 20358. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes. Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Ghibu, S. Mechanics Insights of Alpha-Lipoic Acid against Cardiovascular Diseases during COVID-19 Infection. Int. J. Mol. Sci. 2021, 22, 7979. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Wan, R.; Srikaram, P.; Guntupalli, V.; Hu, C.; Chen, Q.; Gao, P. Cellular senescence in asthma: From pathogenesis to therapeutic challenges. eBioMedicine 2023, 94, 104717. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell cycle arrest is not senescence. Aging 2011, 3, 94–101. [Google Scholar] [CrossRef]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]


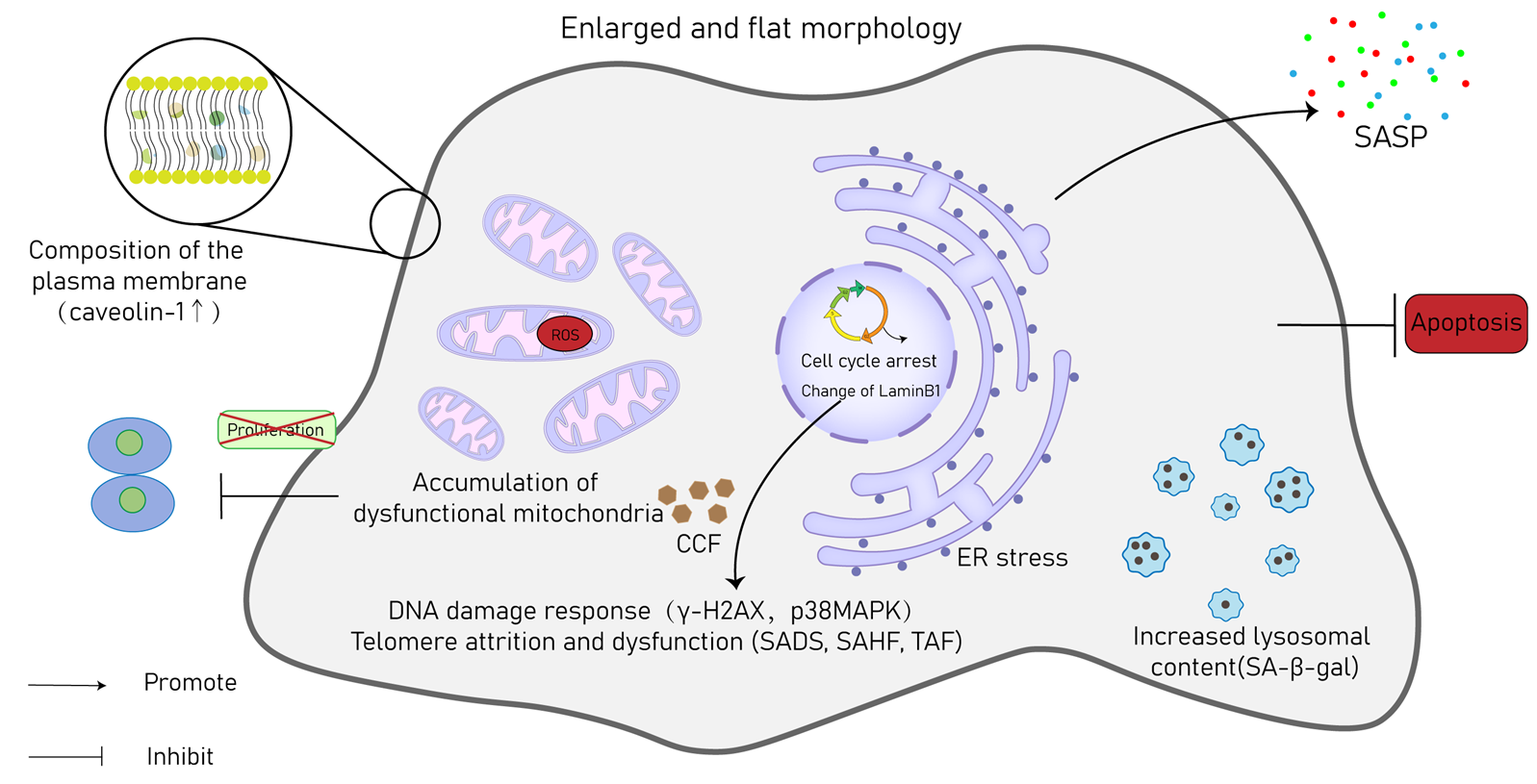

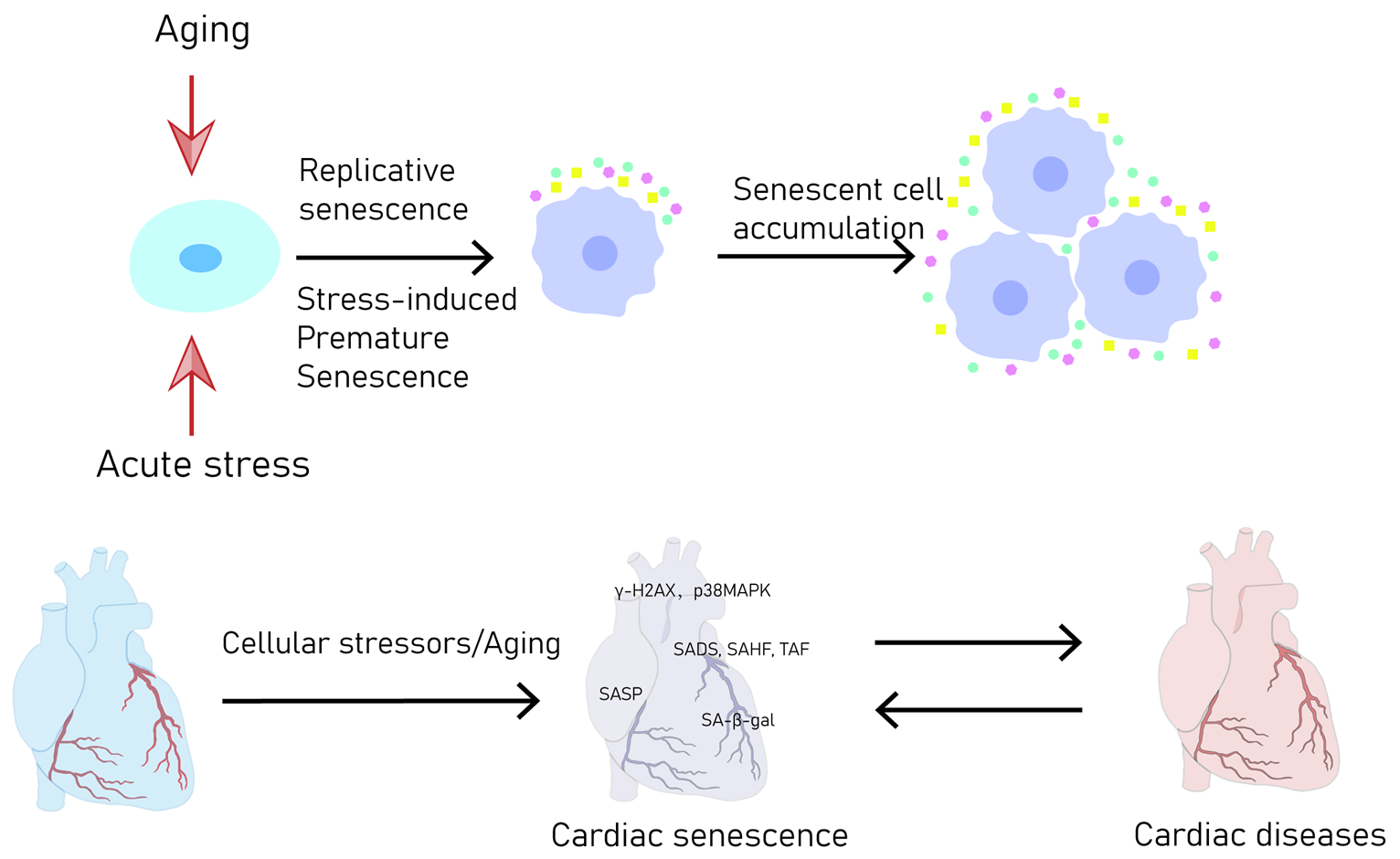{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Initiators | Pathology | Diseases | Hallmarks |
|---|---|---|---|---|
| Cardiomyocytes | Metabolic dysfunction Telomeric shortening Epigenetic factors SASP | Impaired contractility Abnormal conduction patterns Fibrosis | Heart failure Cardiomyopathy Ischemic heart disease Arrhythmias | SA-β-gal, p16, Telomerase, TRF1/2, p53, p27, γ-H2AX, Telomere length, SASP, TAF |
| Endothelial cells | Oxidative stress Vascular inflammation Metabolic factors Epigenetic regulation | Vascular dysfunction Fibronectin accumulation Vascular inflammation Impaired vasodilation | Atherosclerosis HFpEF Pulmonary hypertension Peripheral artery disease Ischemic heart disease | SA-β-gal activity, p53, p21, Cell morphology, p16, SASP, γ-H2AX, ATM, Chk2, Telomerase activity |
| VSMCs | Telomeric shortening DNA damage Oxidative stress Autophagic dysfunction | Inducing local inflammation Impaired smooth muscle Contraction | Atherosclerosis Pulmonary hypertension Hypertension Aortic aneurysm Aortic dissection Myocardial infarction | Cell morphology, SA-β-gal activity, p53, p21, p16, SASP, p27, γ-H2AX, ATM, TRF2, Telomere length, |
| Fibroblasts | Oxidative stress Hypoxia | Limits collagen expression Prevent excessive fibrosis | Heart failure Cardiomyopathy Arrhythmias | SA-β-gal activity, p53, SASP, p21, p16, p19, γ-H2AXCell morphology |
| Immune cells | Telomeric shortening Cell debris ROS | Inflammation Impaired cardiac electrical Conduction | Atherosclerosis Heart failure Arrhythmias | Flow cytometry, CD8+CD57+ T cells, CD28-T cells, D4+CD57+ T cells, CD8+CD28- T cells |
| CPCs | Aging | Fibrotic remodeling Proinflammatory | Ischemic heart diseases | SASP, SA-β-gal, p16, γ-H2AX, Telomere length |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Li, Y.; Ding, H.; Wang, Y.; Xie, Y.; Zhang, X. Cellular Senescence in Cardiovascular Diseases: From Pathogenesis to Therapeutic Challenges. J. Cardiovasc. Dev. Dis. 2023, 10, 439. https://doi.org/10.3390/jcdd10100439
Li D, Li Y, Ding H, Wang Y, Xie Y, Zhang X. Cellular Senescence in Cardiovascular Diseases: From Pathogenesis to Therapeutic Challenges. Journal of Cardiovascular Development and Disease. 2023; 10(10):439. https://doi.org/10.3390/jcdd10100439
Chicago/Turabian StyleLi, Dan, Yongnan Li, Hong Ding, Yuqin Wang, Yafei Xie, and Xiaowei Zhang. 2023. "Cellular Senescence in Cardiovascular Diseases: From Pathogenesis to Therapeutic Challenges" Journal of Cardiovascular Development and Disease 10, no. 10: 439. https://doi.org/10.3390/jcdd10100439