
Increased Infiltration of Extra-Cardiac Cells in Myxomatous Valve Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Gene Targeted Mice
2.2. Histology, Immunohistochemistry/Immunofluorescence
2.3. Western Blotting
2.4. Statistics
3. Results and Discussion
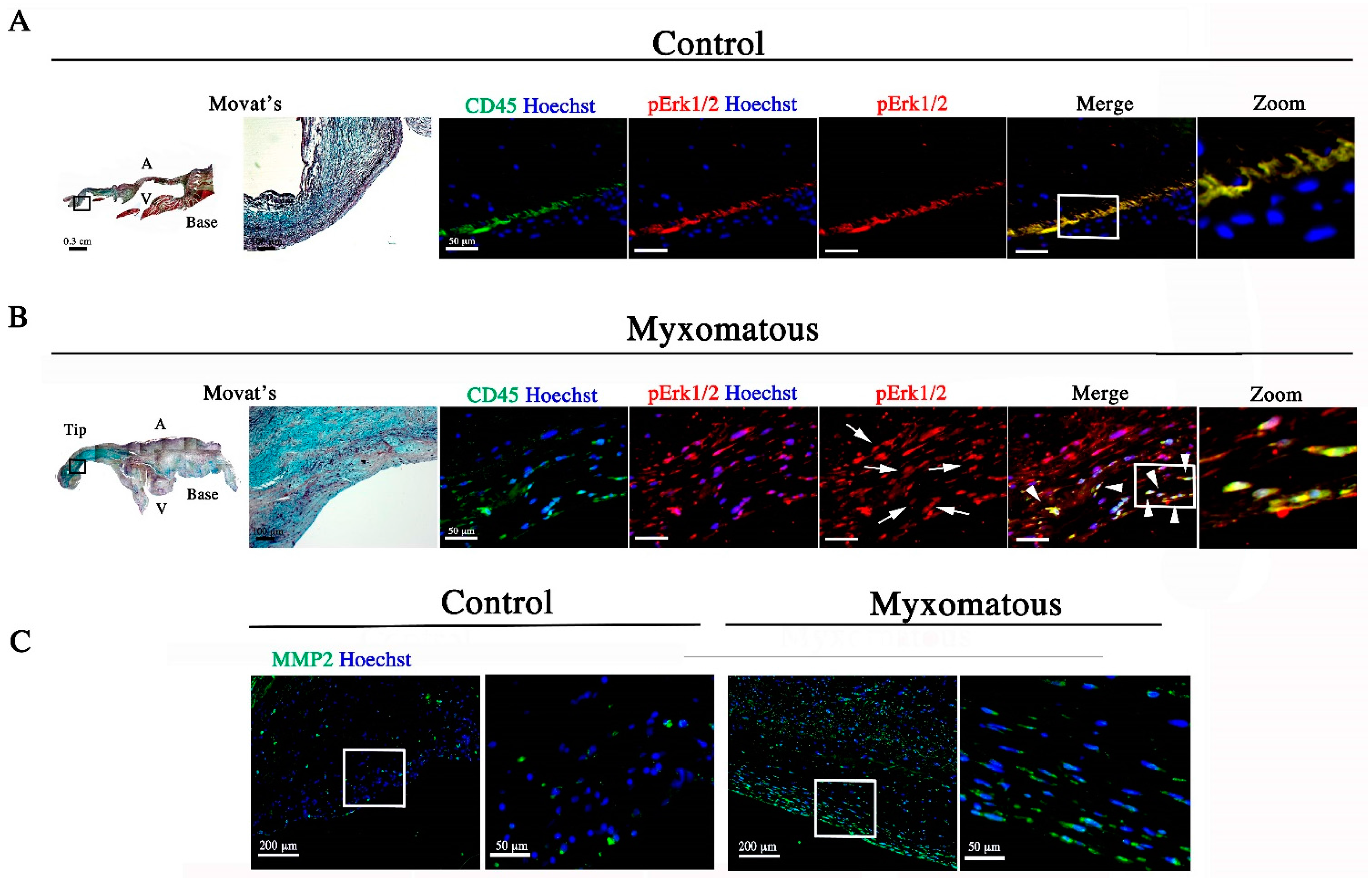
3.1. Adolescent Filamin-A cKO Mice Exhibit Myxomatous Changes by Two Months
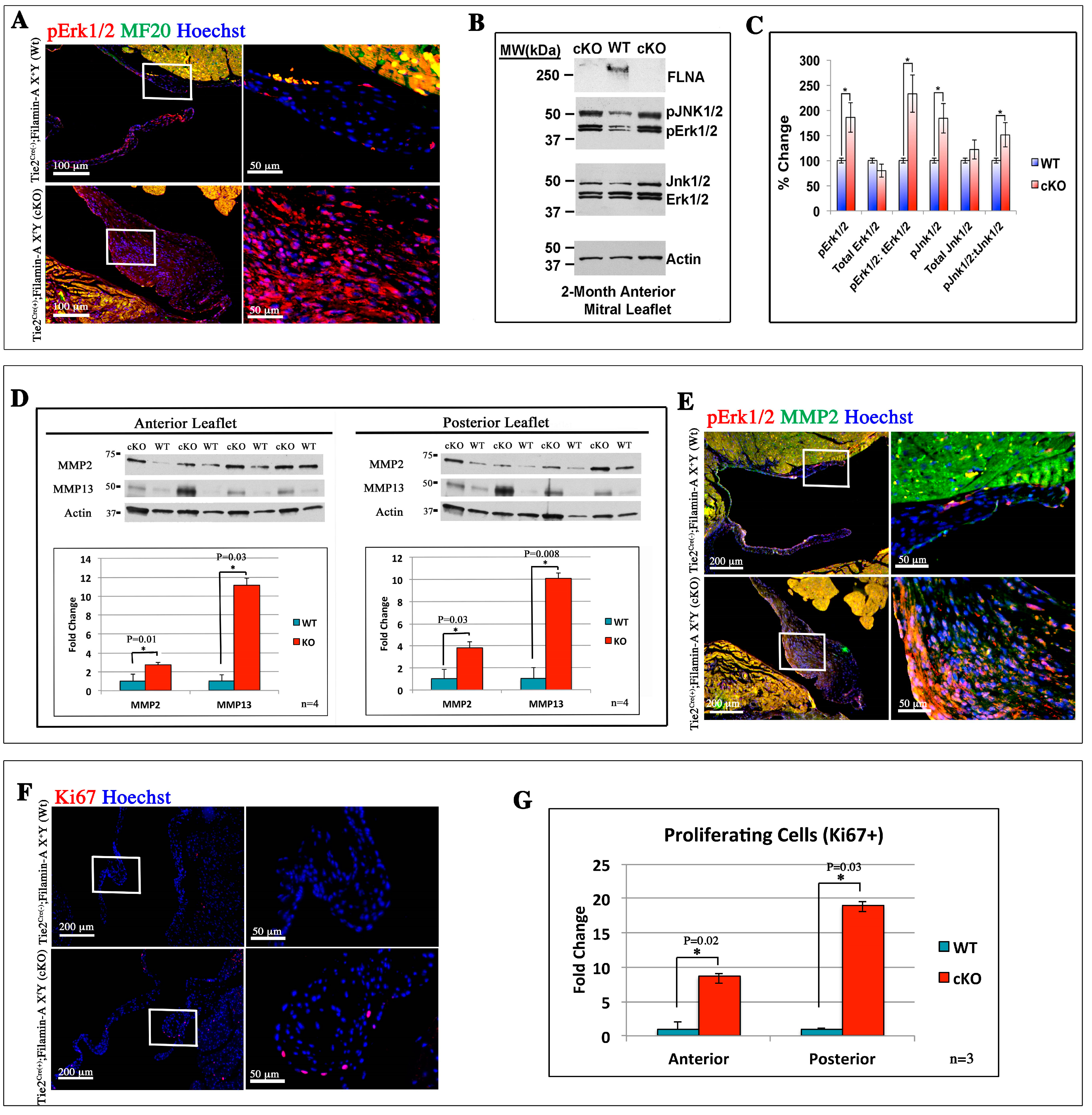
3.2. Filamin-A-Deficient Mice Exhibit Increased Erk Signaling Concurrent with Increased MMP Expression and Cell Proliferation

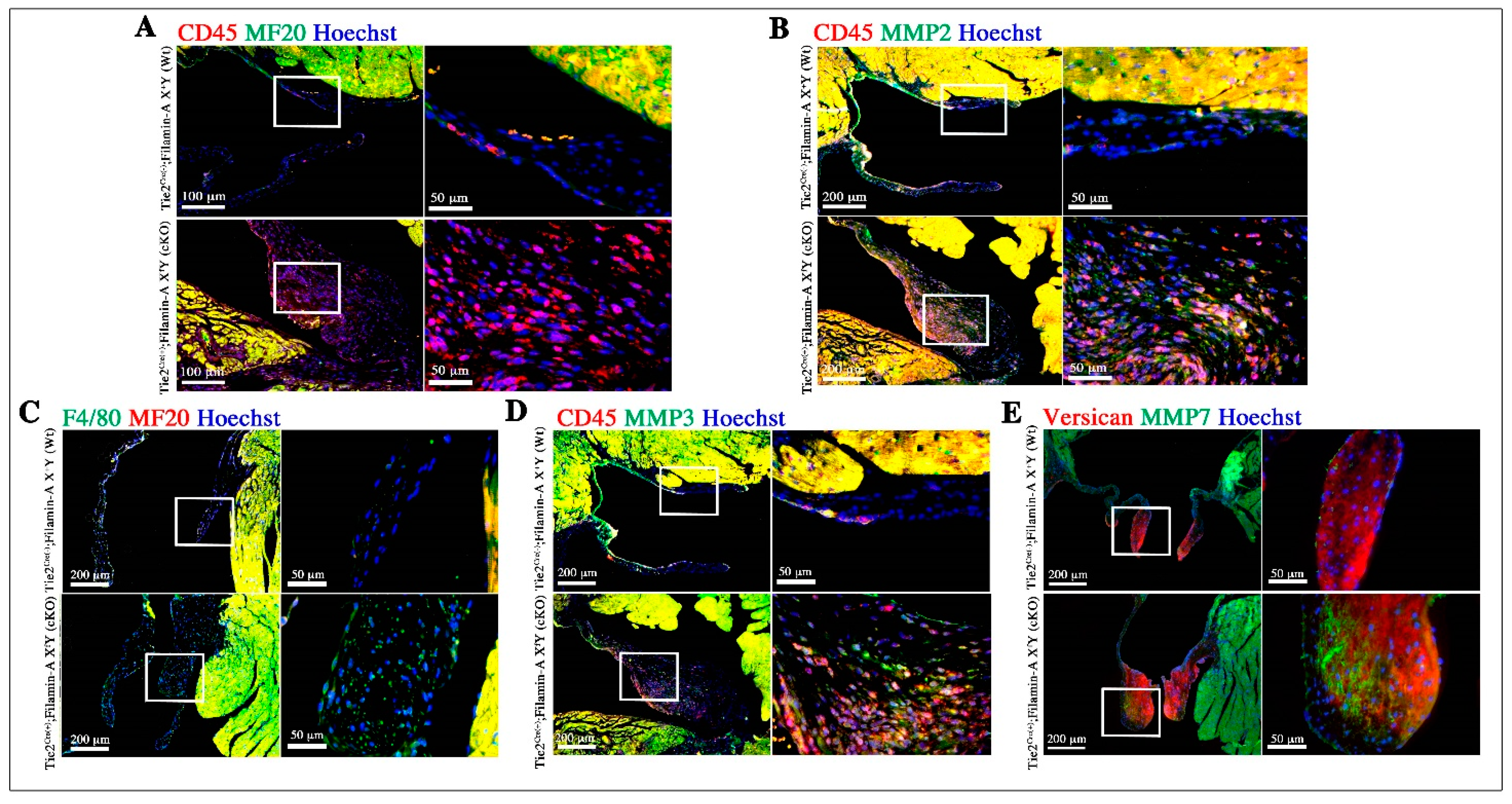
3.3. Increased Infiltration of Hematopoietic-Derived Cells in Myxomatous Valves Contribute to Disease


3.4. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, A.X.; Hartwig, J.H.; Akyurek, L.M. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010, 20, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Sengupta, A.; Glogauer, M.; McCulloch, C.A. Filamin A regulates cell spreading and survival via β1 integrins. Exp. Cell Res. 2008, 314, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Masuda, Y.; Ohta, Y.; Ikeda, K.; Watanabe, K. Filamin associates with Smads and regulates transforming growth factor-β signaling. J. Biol. Chem. 2001, 276, 17871–17877. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.S.; Grundl, M.; Allen, J.S., III; Matter, M.L. R-Ras interacts with filamin a to maintain endothelial barrier function. J. Cell. Physiol. 2011, 226, 2287–2296. [Google Scholar]
- Hart, A.W.; Morgan, J.E.; Schneider, J.; West, K.; McKie, L.; Bhattacharya, S.; Jackson, I.J.; Cross, S.H. Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum. Mol. Genet. 2006, 15, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Chen, M.H.; Moskowitz, I.P.; Mendonza, A.M.; Vidali, L.; Nakamura, F.; Kwiatkowski, D.J.; Walsh, C.A. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19836–19841. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.A.; Moreno-Rodriguez, R.; Wessels, A.; Merot, J.; Bruneval, P.; Chester, A.H.; Yacoub, M.H.; Hagège, A.; Slaugenhaupt, S.A.; Aikawa, E.; et al. Expression of the familial cardiac valvular dystrophy gene, filamin-A, during heart morphogenesis. Dev. Dyn. 2010, 239, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Sauls, K.; de Vlaming, A.; Harris, B.S.; Williams, K.; Wessels, A.; Levine, R.A.; Slaugenhaupt, S.A.; Goodwin, R.L.; Pavone, L.M.; Merot, J.; et al. Developmental basis for filamin-A-associated myxomatous mitral valve disease. Cardiovasc. Res. 2012, 96, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Kisanuki, Y.Y.; Hammer, R.E.; Miyazaki, J.; Williams, S.C.; Richardson, J.A.; Yanagisawa, M. Tie2-Cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 2001, 230, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Chang, H.C.; Leu, T.H.; Maa, M.C.; Hung, W.C. Src oncogene activates MMP-2 expression via the ERK/Sp1 pathway. J. Cell. Physiol. 2006, 207, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Nakayama, K.; Yamamoto, T.; Nishida, E. Regulatory mechanisms and function of ERK MAP kinases. J. Biochem. 2004, 136, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Hajdu, Z.; Romeo, S.J.; Fleming, P.A.; Markwald, R.R.; Visconti, R.P.; Drake, C.J. Recruitment of bone marrow-derived valve interstitial cells is a normal homeostatic process. J. Mol. Cell. Cardiol. 2011, 51, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Visconti, R.P.; Ebihara, Y.; LaRue, A.C.; Fleming, P.A.; McQuinn, T.C.; Masuya, M.; Minamiguchi, H.; Markwald, R.R.; Ogawa, M.; Drake, C.J. An in vivo analysis of hematopoietic stem cell potential: Hematopoietic origin of cardiac valve interstitial cells. Circ. Res. 2006, 98, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Zhou, Z.; Schadler, K.; Jia, S.F.; Kleinerman, E.S. Bone marrow subsets differentiate into endothelial cells and pericytes contributing to Ewing's tumor vessels. Mol. Cancer Res. 2008, 6, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kisseleva, T. Bone marrow-derived fibrocytes contribute to liver fibrosis. Exp. Biol. Med. 2015, 240, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.M.; Cheng, A.; Myers, L.A.; Martinez-Murillo, F.; Jie, C.; Bedja, D.; Gabrielson, K.L.; Hausladen, J.M.; Mecham, R.P.; Judge, D.P.; et al. TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J. Clin. Investig. 2004, 114, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Dugan, S.L.; Temme, R.T.; Olson, R.A.; Mikhailov, A.; Law, R.; Mahmood, H.; Noor, A.; Vincent, J.B. New recessive truncating mutation in LTBP3 in a family with oligodontia, short stature, and mitral valve prolapse. Am. J. Med. Genet. A 2015, 167, 1396–1399. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.J.; Doyle, J.J.; Bessling, S.L.; Maragh, S.; Lindsay, M.E.; Schepers, D.; Gillis, E.; Mortier, G.; Homfray, T.; Sauls, K.; et al. Mutations in the TGF-β repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat. Genet. 2012, 44, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Malev, E.G.; Zemtsovskiĭ, E.V.; Omel’chenko, M.I.; Vasina, L.V. The role of transforming growth factor-β in the pathogenesis of mitral valve prolapse. Kardiologiia 2012, 52, 34–39. [Google Scholar] [PubMed]
- Boileau, C.; Guo, D.C.; Hanna, N.; Regalado, E.S.; Detaint, D.; Gong, L.; Varret, M.; Prakash, S.K.; Li, A.H.; d’Indy, H.; Braverman, A.C.; et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat. Genet. 2012, 44, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.W.; Shi, G.P.; Kuzuya, M.; Sasaki, T.; Okumura, K.; Murohara, T. Role for cysteine protease cathepsins in heart disease: Focus on biology and mechanisms with clinical implication. Circulation 2012, 125, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.W.; Huang, Z.; Kuzuya, M.; Okumura, K.; Murohara, T. Cysteine protease cathepsins in atherosclerosis-based vascular disease and its complications. Hypertension 2011, 58, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Cheng, X.W.; Shi, G.P.; Hu, L.; Inoue, A.; Yamamura, Y.; Wu, H.; Takeshita, K.; Li, X.; Huang, Z.; et al. Cathepsin K-mediated Notch1 activation contributes to neovascularization in response to hypoxia. Nat. Commun. 2014, 5, 3838. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.J.; Koster, H.; Moosdorf, R. CD34+ fibrocytes in normal mitral valves and myxomatous mitral valve degeneration. Pathol. Res. Pract. 2005, 201, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Geirsson, A.; Singh, M.; Ali, R.; Abbas, H.; Li, W.; Sanchez, J.A.; Hashim, S.; Tellides, G. Modulation of transforming growth factor-β signaling and extracellular matrix production in myxomatous mitral valves by angiotensin II receptor blockers. Circulation 2012, 126, S189–S197. [Google Scholar] [CrossRef] [PubMed]
- Han, R.I.; Black, A.; Culshaw, G.J.; French, A.T.; Else, R.W.; Corcoran, B.M. Distribution of myofibroblasts, smooth muscle-like cells, macrophages, and mast cells in mitral valve leaflets of dogs with myxomatous mitral valve disease. Am. J. Vet. Res. 2008, 69, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Zois, N.E.; Moesgaard, S.G.; Kjelgaard-Hansen, M.; Rasmussen, C.E.; Falk, T.; Fossing, C.; Häggström, J.; Pedersen, H.D.; Olsen, L.H. Circulating cytokine concentrations in dogs with different degrees of myxomatous mitral valve disease. Vet. J. 2012, 192, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, I. Extracellular matrix remodelling: The role of matrix metalloproteinases. J. Pathol. 2003, 200, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Ploeger, D.T.; Hosper, N.A.; Schipper, M.; Koerts, J.A.; de Rond, S.; Bank, R.A. Cell plasticity in wound healing: Paracrine factors of M1/M2 polarized macrophages influence the phenotypical state of dermal fibroblasts. Cell Commun. Signal. 2013, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Howe, L.R.; Falcone, D.J. Extracellular matrix-induced cyclooxygenase-2 regulates macrophage proteinase expression. J. Biol. Chem. 2004, 279, 22039–22046. [Google Scholar] [CrossRef] [PubMed]
- Togashi, M.; Tamura, K.; Nitta, T.; Ishizaki, M.; Sugisaki, Y.; Fukuda, Y. Role of matrix metalloproteinases and their tissue inhibitor of metalloproteinases in myxomatous change of cardiac floppy valves. Pathol. Int. 2007, 57, 251–259. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sauls, K.; Toomer, K.; Williams, K.; Johnson, A.J.; Markwald, R.R.; Hajdu, Z.; Norris, R.A. Increased Infiltration of Extra-Cardiac Cells in Myxomatous Valve Disease. J. Cardiovasc. Dev. Dis. 2015, 2, 200-213. https://doi.org/10.3390/jcdd2030200
Sauls K, Toomer K, Williams K, Johnson AJ, Markwald RR, Hajdu Z, Norris RA. Increased Infiltration of Extra-Cardiac Cells in Myxomatous Valve Disease. Journal of Cardiovascular Development and Disease. 2015; 2(3):200-213. https://doi.org/10.3390/jcdd2030200
Chicago/Turabian StyleSauls, Kimberly, Katelynn Toomer, Katherine Williams, Amanda J. Johnson, Roger R. Markwald, Zoltan Hajdu, and Russell A. Norris. 2015. "Increased Infiltration of Extra-Cardiac Cells in Myxomatous Valve Disease" Journal of Cardiovascular Development and Disease 2, no. 3: 200-213. https://doi.org/10.3390/jcdd2030200
APA StyleSauls, K., Toomer, K., Williams, K., Johnson, A. J., Markwald, R. R., Hajdu, Z., & Norris, R. A. (2015). Increased Infiltration of Extra-Cardiac Cells in Myxomatous Valve Disease. Journal of Cardiovascular Development and Disease, 2(3), 200-213. https://doi.org/10.3390/jcdd2030200