
Drosophila in the Heart of Understanding Cardiac Diseases: Modeling Channelopathies and Cardiomyopathies in the Fruitfly

Abstract
:1. Introduction
2. Cardiovascular Disease Modeling Using Drosophila
2.1. Channelopathies
2.2. Cardiomyopathies
2.2.1. HCM
2.2.2. DCM
2.2.3. RCM
3. Pathological Mechanisms Investigated
3.1. Impaired Calcium Handling
3.2. Altered Metabolism
3.3. Increased Oxidative Stress and Mitochondrial Dysfunction
3.4. Remodeling of Extracellular Matrix
4. Challenges in CVDs Modeling
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nichols, M.; Townsend, N.; Scarborough, P.; Rayner, M. Cardiovascular disease in Europe 2014: Epidemiological update. Eur. Heart J. 2014, 35, 2929. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Executive summary: Heart disease and stroke statistics—2014 update: A report from the American Heart Association. Circulation 2014, 129, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Jensen, B.C.; Weck, K.E.; Willis, M.S. Evolving molecular diagnostics for familial cardiomyopathies: At the heart of it all. Expert Rev. Mol. Diagn. 2010, 10, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002, 287, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Dec, G.W.; Fuster, V. Idiopathic dilated cardiomyopathy. N. Engl. J. Med. 1994, 331, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Codd, M.B.; Sugrue, D.D.; Gersh, B.J.; Melton, L.J., 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 1989, 80, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, S.S.; Fallon, J.T.; Fuster, V. Restrictive cardiomyopathy. N. Engl. J. Med. 1997, 336, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Gimeno, J.R.; Bahl, A.; Steffensen, U.; Steffensen, M.; Osman, E.; Thaman, R.; Mogensen, J.; Elliott, P.M.; Doi, Y.; et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J. Am. Coll. Cardiol. 2007, 49, 2419–2426. [Google Scholar] [CrossRef] [PubMed]
- Sen-Chowdhry, S.; Morgan, R.D.; Chambers, J.C.; McKenna, W.J. Arrhythmogenic cardiomyopathy: Etiology, diagnosis, and treatment. Annu. Rev. Med. 2010, 61, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Corrado, D.; Thiene, G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol. Rev. 1999, 7, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Tabib, A.; Loire, R.; Chalabreysse, L.; Meyronnet, D.; Miras, A.; Malicier, D.; Thivolet, F.; Chevalier, P.; Bouvagnet, P. Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 2003, 108, 3000–3005. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. Arrhythmogenic right ventricular cardiomyopathy: A paradigm of overlapping disorders. Ann. Noninvasive Electrocardiol. 2008, 13, 325–326. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Bedell, M.A.; Largaespada, D.A.; Jenkins, N.A.; Copeland, N.G. Mouse models of human disease. Part II: Recent progress and future directions. Genes Dev. 1997, 11, 11–43. [Google Scholar] [CrossRef] [PubMed]
- Peters, L.L.; Robledo, R.F.; Bult, C.J.; Churchill, G.A.; Paigen, B.J.; Svenson, K.L. The mouse as a model for human biology: A resource guide for complex trait analysis. Nat. Rev. Genet. 2007, 8, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Martins, P.C.M.; Ayub-Guerrieri, D.; Martins-Bach, A.B.; Onofre-Oliveira, P.; Malheiros, J.M.; Tannus, A.; de Sousa, P.L.; Carlier, P.G.; Vainzof, M. Dmdmdx/Largemyd: A new mouse model of neuromuscular diseases useful for studying physiopathological mechanisms and testing therapies. Dis. Model. Mech. 2013, 6, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Bier, E.; Bodmer, R. Drosophila, an emerging model for cardiac disease. Gene 2004, 342, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.; Hughes, S.M.; Segalat, L. Other model organisms for sarcomeric muscle diseases. Adv. Exp. Med. Biol. 2008, 642, 192–206. [Google Scholar] [PubMed]
- Lloyd, T.E.; Taylor, J.P. Flightless flies: Drosophila models of neuromuscular disease. Ann. N. Y. Acad. Sci. 2010, 1184, e1–e20. [Google Scholar] [CrossRef] [PubMed]
- Piazza, N.; Wessells, R.J. Drosophila Models of Cardiac Disease. Prog. Mol. Biol. Transl. Sci. 2011, 100, 155–210. [Google Scholar] [PubMed]
- Plantié, E.; Migocka-Patrzałek, M.; Daczewska, M.; Jagla, K. Model Organisms in the Fight against Muscular Dystrophy: Lessons from Drosophila and Zebrafish. Molecules 2015, 20, 6237–6253. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development 1993, 118, 719–729. [Google Scholar] [PubMed]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Black, B.L. Transcriptional pathways in second heart field development. Semin. Cell Dev. Biol. 2007, 18, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Mohapatra, B.; Akasaka, T.; Liu, J.; Ocorr, K.; Towbin, J.A.; Bodmer, R. Transcription factor neuromancer/TBX20 is required for cardiac function in Drosophila with implications for human heart disease. Proc. Natl. Acad. Sci. USA 2008, 105, 19833–19838. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Bodmer, R. Partial loss of GATA factor Pannier impairs adult heart function in Drosophila. Hum. Mol. Genet. 2009, 18, 3153–3163. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Wythe, J.D.; Liu, J.; Cartry, J.; Vogler, G.; Mohapatra, B.; Otway, R.T.; Huang, Y.; King, I.N.; Maillet, M.; et al. Tinman/Nkx2-5 acts via miR-1 and upstream of Cdc42 to regulate heart function across species. J. Cell Biol. 2011, 193, 1181–1196. [Google Scholar] [CrossRef] [PubMed]
- Jay, P.Y.; Harris, B.S.; Maguire, C.T.; Buerger, A.; Wakimoto, H.; Tanaka, M.; Kupershmidt, S.; Roden, D.M.; Schultheiss, T.M.; O’Brien, T.X.; et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J. Clin. Investig. 2004, 113, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, I.P.G.; Kim, J.B.; Moore, M.L.; Wolf, C.M.; Peterson, M.A.; Shendure, J.; Nobrega, M.A.; Yokota, Y.; Berul, C.; Izumo, S.; et al. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell 2007, 129, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, R. Heart development in Drosophila and its relationship to vertebrates. Trends Cardiovasc. Med. 1995, 5, 21–28. [Google Scholar] [CrossRef]
- Bodmer, R.; Venkatesh, T.V. Heart development in Drosophila and vertebrates: Conservation of molecular mechanisms. Dev. Genet. 1998, 22, 181–186. [Google Scholar] [CrossRef]
- Cripps, R.M.; Olson, E.N. Control of cardiac development by an evolutionarily conserved transcriptional network. Dev. Biol. 2002, 246, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Sink, H. Muscle Development in Drosophilia; Springer Science & Business Media: New York, NY, USA, 2007. [Google Scholar]
- Choma, M.A.; Suter, M.J.; Vakoc, B.J.; Bouma, B.E.; Tearney, G.J. Physiological homology between Drosophila melanogaster and vertebrate cardiovascular systems. Dis. Model. Mech. 2011, 4, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Ahrens, C.H.; Alayari, N.N.; Qeli, E.; Rucker, J.; Reedy, M.C.; Zmasek, C.M.; Gucek, M.; Cole, R.N.; Van Eyk, J.E.; et al. A mighty small heart: The cardiac proteome of adult Drosophila melanogaster. PLoS ONE 2011, 6, e18497. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Ringo, J.; Dowse, H. Dynamin, encoded by shibire, is central to cardiac function. J. Exp. Zool. 2001, 289, 81–89. [Google Scholar] [CrossRef]
- Papaefthmiou, C.; Theophilidis, G. An in vitro method for recording the electrical activity of the isolated heart of the adult Drosophila melanogaster. In Vitro Cell. Dev. Biol. Anim. 2001, 37, 445–449. [Google Scholar] [CrossRef]
- Lalevée, N.; Monier, B.; Sénatore, S.; Perrin, L.; Sémériva, M. Control of Cardiac Rhythm by ORK1, a Drosophila Two-Pore Domain Potassium Channel. Curr. Biol. 2006, 16, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Dulcis, D.; Levine, R.B.; Ewer, J. Role of the neuropeptide CCAP in Drosophila cardiac function. J. Neurobiol. 2005, 64, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Wasserthal, L.T. Drosophila flies combine periodic heartbeat reversal with a circulation in the anterior body mediated by a newly discovered anterior pair of ostial valves and “venous” channels. J. Exp. Biol. 2007, 210, 3707–3719. [Google Scholar] [CrossRef] [PubMed]
- Dowse, H.; Ringo, J.; Power, J.; Johnson, E.; Kinney, K.; White, L. A congenital heart defect in Drosophila caused by an action-potential mutation. J. Neurogenet. 1995, 10, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Ringo, J.; Bray, N.; Dowse, H. Genetic and Pharmacological Identification of Ion Channels Central to the Drosophila Cardiac Pacemaker. J. Neurogenet. 1998, 12, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Wessells, R.J.; Bodmer, R. Screening assays for heart function mutants in Drosophila. BioTechniques 2004, 37, 58–66. [Google Scholar] [PubMed]
- Paternostro, G.; Vignola, C.; Bartsch, D.-U.; Omens, J.H.; McCulloch, A.D.; Reed, J.C. Age-Associated Cardiac Dysfunction in Drosophila melanogaster. Circ. Res. 2001, 88, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, A.D.; Paternostro, G. Cardiac systems biology. Ann. N. Y. Acad. Sci. 2005, 1047, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Ocorr, K.; Reeves, N.L.; Wessells, R.J.; Fink, M.; Chen, H.-S.V.; Akasaka, T.; Yasuda, S.; Metzger, J.M.; Giles, W.; Posakony, J.W.; et al. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc. Natl. Acad. Sci. USA 2007, 104, 3943–3948. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.; Callol-Massot, C.; Chu, A.; Ruiz-Lozano, P.; Belmonte, J.; Giles, W.; Bodmer, R.; Ocorr, K. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. BioTechniques 2009, 46, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Amrein, H.; Izatt, J.A.; Choma, M.A.; Reedy, M.C.; Rockman, H.A. Drosophila as a model for the identification of genes causing adult human heart disease. Proc. Natl. Acad. Sci. USA 2006, 103, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.; Li, A.; Tanzi, R.E.; Zhou, C. Optogenetic pacing in Drosophila melanogaster. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, G.; Zambon, A.C.; Fuhrmann, A.; Bernstein, S.I.; Bodmer, R.; Engler, A.J.; Cammarato, A. Measuring passive myocardial stiffness in Drosophila melanogaster to investigate diastolic dysfunction. J. Cell. Mol. Med. 2012, 16, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Klinedinst, S.; Ocorr, K.; Bustamante, E.L.; Kim, S.K.; Bodmer, R. The ATP-sensitive potassium (KATP) channel-encoded dSUR gene is required for Drosophila heart function and is regulated by tinman. Proc. Natl. Acad. Sci. USA 2006, 103, 11999–12004. [Google Scholar] [CrossRef] [PubMed]
- Frolov, R.V.; Singh, S. Inhibition of ion channels and heart beat in Drosophila by selective COX-2 inhibitor SC-791. PLoS ONE 2012, 7, e38759. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Dambacher, C.M.; Knowles, A.F.; Kronert, W.A.; Bodmer, R.; Ocorr, K.; Bernstein, S.I. Myosin transducer mutations differentially affect motor function, myofibril structure, and the performance of skeletal and cardiac muscles. Mol. Biol. Cell 2008, 19, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Taghli-Lamallem, O.; Akasaka, T.; Hogg, G.; Nudel, U.; Yaffe, D.; Chamberlain, J.S.; Ocorr, K.; Bodmer, R. Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell 2008, 7, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Daniels, J.; Glaser, A.E.; Wolf, M.J. Raf-mediated cardiac hypertrophy in adult Drosophila. Dis. Model. Mech. 2013, 6, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Kaushik, G.; Engler, A.J.; Lehman, W.; Cammarato, A. A Drosophila melanogaster Model of Diastolic Dysfunction and Cardiomyopathy Based on Impaired Troponin-T Function. Circ. Res. 2014, 114, e6–e17. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Daniels, J.P.; Wu, H.; Wolf, M.J. Cardiac hypertrophy induced by active Raf depends on Yorkie-mediated transcription. Sci. Signal. 2015, 8, ra13. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.E.; Yu, L.; Wolf, M.J.; Rockman, H.A. Galactokinase is a novel modifier of calcineurin-induced cardiomyopathy in Drosophila. Genetics 2014, 198, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Kamb, A.; Iverson, L.E.; Tanouye, M.A. Molecular characterization of Shaker, a Drosophila gene that encodes a potassium channel. Cell 1987, 50, 405–413. [Google Scholar] [CrossRef]
- Papazian, D.M.; Schwarz, T.L.; Tempel, B.L.; Jan, Y.N.; Jan, L.Y. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science 1987, 237, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Tempel, B.L.; Papazian, D.M.; Schwarz, T.L.; Jan, Y.N.; Jan, L.Y. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science 1987, 237, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, D.R.; Pond, A.L.; McCarthy, P.M.; Trimmer, J.S.; Nerbonne, J.M. Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ. Res. 1997, 80, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Li, G.R.; Dong, M.Q. Pharmacology of cardiac potassium channels. Adv. Pharmacol. 2010, 59, 93–134. [Google Scholar] [PubMed]
- Butler, A.; Wei, A.G.; Baker, K.; Salkoff, L. A family of putative potassium channel genes in Drosophila. Science 1989, 243, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, M.; Wei, A.A.; Salkoff, L. Shaker, Shal, Shab, and Shaw express independent K+ current systems. Neuron 1991, 7, 763–773. [Google Scholar] [CrossRef]
- Tsunoda, S.; Salkoff, L. Genetic analysis of Drosophila neurons: Shal, Shaw, and Shab encode most embryonic potassium currents. J. Neurosci. 1995, 15, 1741–1754. [Google Scholar] [PubMed]
- Roberds, S.L.; Tamkun, M.M. Cloning and tissue-specific expression of five voltage-gated potassium channel cDNAs expressed in rat heart. Proc. Natl. Acad. Sci. USA 1991, 88, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barry, D.M.; Li, H.; Brunet, S.; Guo, W.; Nerbonne, J.M. Attenuation of the slow component of delayed rectification, action potential prolongation, and triggered activity in mice expressing a dominant-negative Kv2 alpha subunit. Circ. Res. 1999, 85, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Ordog, B.; Brutyo, E.; Puskas, L.G.; Papp, J.G.; Varro, A.; Szabad, J.; Boldogkoi, Z. Gene expression profiling of human cardiac potassium and sodium channels. Int. J. Cardiol. 2006, 111, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, N.; Wichter, T.; Varro, A.; Szuts, V.; Lamirault, G.; Eckardt, L.; Paul, M.; Breithardt, G.; Schulze-Bahr, E.; Escande, D.; et al. Transcriptional profiling of ion channel genes in Brugada syndrome and other right ventricular arrhythmogenic diseases. Eur. Heart J. 2009, 30, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ye, D.; Tester, D.J.; Crotti, L.; Mugione, A.; Nesterenko, V.V.; Albertson, R.M.; Antzelevitch, C.; Schwartz, P.J.; Ackerman, M.J. Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 2011, 8, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Drysdale, R.; Warmke, J.; Kreber, R.; Ganetzky, B. Molecular characterization of eag: A gene affecting potassium channels in Drosophila melanogaster. Genetics 1991, 127, 497–505. [Google Scholar] [PubMed]
- Warmke, J.W.; Ganetzky, B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. USA 1994, 91, 3438–3442. [Google Scholar] [CrossRef] [PubMed]
- Titus, S.A.; Warmke, J.W.; Ganetzky, B. The Drosophila erg K+ channel polypeptide is encoded by the seizure locus. J. Neurosci. 1997, 17, 875–881. [Google Scholar] [PubMed]
- January, C.T.; Gong, Q.; Zhou, Z. Long QT syndrome: Cellular basis and arrhythmia mechanism in LQT2. J. Cardiovasc. Electrophysiol. 2000, 11, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.S.; Robertson, G.A.; Ganetzky, B. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science 1991, 253, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Pallanck, L.; Ganetzky, B. Cloning and characterization of human and mouse homologs of the Drosophila calcium-activated potassium channel gene, slowpoke. Hum. Mol. Genet. 1994, 3, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Brenner, R.; Perez, G.J.; Bonev, A.D.; Eckman, D.M.; Kosek, J.C.; Wiler, S.W.; Patterson, A.J.; Nelson, M.T.; Aldrich, R.W. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 2000, 407, 870–876. [Google Scholar] [PubMed]
- Fernandez-Fernandez, J.M.; Tomas, M.; Vazquez, E.; Orio, P.; Latorre, R.; Senti, M.; Marrugat, J.; Valverde, M.A. Gain-of-function mutation in the KCNMB1 potassium channel subunit is associated with low prevalence of diastolic hypertension. J. Clin. Investig. 2004, 113, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. J. Physiol. 2000, 525, Pt 2. 285–298. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M.; Kass, R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005, 85, 1205–1253. [Google Scholar] [CrossRef] [PubMed]
- Vincent, G.M.; Timothy, K.; Fox, J.; Zhang, L. The inherited long QT syndrome: From ion channel to bedside. Cardiol. Rev. 1999, 7, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C. Dysfunction of delayed rectifier potassium channels in an inherited cardiac arrhythmia. Ann. N. Y. Acad. Sci. 1999, 868, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Moss, A.J. Long QT syndrome. J. Am. Coll. Cardiol. 2008, 51, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Ocorr, K. Drug discovery through functional screening in the Drosophila heart. Methods Mol. Biol. 2009, 577, 235–249. [Google Scholar] [PubMed]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: The aging heart in health: Links to heart disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Ocorr, K.; Bodmer, R.; Cartry, J. Drosophila as a model to study cardiac aging. Exp. Gerontol. 2011, 46, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M. Studying cardiac arrhythmias in the mouse—A reasonable model for probing mechanisms? Trends Cardiovasc. Med. 2004, 14, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Sasaki, N.; Miki, T.; Sakamoto, N.; Ohmoto-Sekine, Y.; Tamagawa, M.; Seino, S.; Marban, E.; Nakaya, H. Role of sarcolemmal K(ATP) channels in cardioprotection against ischemia/reperfusion injury in mice. J. Clin. Investig. 2002, 109, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ke, L.; Mackovicova, K.; Der Want, J.J.L.V.; Sibon, O.C.M.; Tanguay, R.M.; Morrow, G.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J.J.M. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell. Cardiol. 2011, 51, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.J.; Shiroshita-Takeshita, A.; Qi, X.; Yeh, Y.H.; Chartier, D.; van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Nattel, S. Induction of heat shock response protects the heart against atrial fibrillation. Circ. Res. 2006, 99, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Hypertrophic cardiomyopathy: From genetics to treatment. Eur. J. Clin. Investig. 2010, 40, 360–369. [Google Scholar] [CrossRef]
- Seidman, J.G.; Seidman, C. The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 2001, 104, 557–567. [Google Scholar] [CrossRef]
- Alcalai, R.; Seidman, J.G.; Seidman, C.E. Genetic basis of hypertrophic cardiomyopathy: From bench to the clinics. J. Cardiovasc. Electrophysiol. 2008, 19, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J. Genetic determinants of cardiac hypertrophy. Curr. Opin. Cardiol. 2008, 23, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Geier, C.; Perrot, A.; Ozcelik, C.; Binner, P.; Counsell, D.; Hoffmann, K.; Pilz, B.; Martiniak, Y.; Gehmlich, K.; van der Ven, P.F.; et al. Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation 2003, 107, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Arimura, T.; Itoh-Satoh, M.; Ueda, K.; Hohda, S.; Inagaki, N.; Takahashi, M.; Hori, H.; Yasunami, M.; Nishi, H.; et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Vasile, V.C.; Will, M.L.; Ommen, S.R.; Edwards, W.D.; Olson, T.M.; Ackerman, M.J. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol. Genet. Metab. 2006, 87, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Weisleder, N.; Batalden, K.B.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Wehrens, X.H.; Claycomb, W.C.; Ko, J.K.; Hwang, M.; et al. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J. Mol. Cell. Cardiol. 2007, 42, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Charron, P.; Carrier, L.; Ledeuil, C.; Cheav, T.; Pichereau, C.; Benaiche, A.; Isnard, R.; Dubourg, O.; Burban, M.; et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003, 107, 2227–2232. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Vasile, V.C.; Ommen, S.R.; Will, M.L.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Lowey, S. Functional consequences of mutations in the myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. Trends Cardiovasc. Med. 2002, 12, 348–354. [Google Scholar] [CrossRef]
- Tardiff, J.C. Sarcomeric proteins and familial hypertrophic cardiomyopathy: Linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail. Rev. 2005, 10, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Tyska, M.J.; Hayes, E.; Giewat, M.; Seidman, C.E.; Seidman, J.G.; Warshaw, D.M. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 2000, 86, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Debold, E.P.; Schmitt, J.P.; Patlak, J.B.; Beck, S.E.; Moore, J.R.; Seidman, J.G.; Seidman, C.; Warshaw, D.M. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H284–H291. [Google Scholar] [CrossRef] [PubMed]
- Sussman, M.A.; Lim, H.W.; Gude, N.; Taigen, T.; Olson, E.N.; Robbins, J.; Colbert, M.C.; Gualberto, A.; Wieczorek, D.F.; Molkentin, J.D. Prevention of cardiac hypertrophy in mice by calcineurin inhibition. Science 1998, 281, 1690–1693. [Google Scholar] [CrossRef] [PubMed]
- Taigen, T.; De Windt, L.J.; Lim, H.W.; Molkentin, J.D. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2000, 97, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef]
- Schonberger, J.; Seidman, C.E. Many roads lead to a broken heart: The genetics of dilated cardiomyopathy. Am. J. Hum. Genet. 2001, 69, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Morales, A.; Siegfried, J.D. Clinical and genetic issues in dilated cardiomyopathy: A review for genetics professionals. Genet. Med. 2010, 12, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; Otway, R.; Richmond, Z. Genetics of dilated cardiomyopathy. Heart Fail. Clin. 2010, 6, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Towbin, J.A. Dilated cardiomyopathy. Lancet 2010, 375, 752–762. [Google Scholar] [CrossRef]
- Morimoto, S.; Lu, Q.W.; Harada, K.; Takahashi-Yanaga, F.; Minakami, R.; Ohta, M.; Sasaguri, T.; Ohtsuki, I. Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2002, 99, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.W.; Morimoto, S.; Harada, K.; Du, C.K.; Takahashi-Yanaga, F.; Miwa, Y.; Sasaguri, T.; Ohtsuki, I. Cardiac troponin T mutation R141W found in dilated cardiomyopathy stabilizes the troponin T-tropomyosin interaction and causes a Ca2+ desensitization. J. Mol. Cell. Cardiol. 2003, 35, 1421–1427. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, G.; Harada, K.; Gomes, A.V.; Kerrick, W.G.; Potter, J.D. Different functional properties of troponin T mutants that cause dilated cardiomyopathy. J. Biol. Chem. 2003, 278, 41670–41676. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.V.; Potter, J.D. Molecular and cellular aspects of troponin cardiomyopathies. Ann. N. Y. Acad. Sci. 2004, 1015, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Melkani, G.C.; Bodmer, R.; Ocorr, K.; Bernstein, S.I. The UNC-45 chaperone is critical for establishing myosin-based myofibrillar organization and cardiac contractility in the Drosophila heart model. PLoS ONE 2011, 6, e22579. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.N.; Potter, J.D. Sarcomeric protein mutations in dilated cardiomyopathy. Heart Fail. Rev. 2005, 10, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Allikian, M.J.; Bhabha, G.; Dospoy, P.; Heydemann, A.; Ryder, P.; Earley, J.U.; Wolf, M.J.; Rockman, H.A.; McNally, E.M. Reduced life span with heart and muscle dysfunction in Drosophila sarcoglycan mutants. Hum. Mol. Genet. 2007, 16, 2933–2943. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Lee, T.; Lin, N.; Wolf, M.J. Affecting Rhomboid-3 function causes a dilated heart in adult Drosophila. PLoS Genet. 2010, 6, e1000969. [Google Scholar] [CrossRef] [PubMed]
- Suter, T.M.; Procter, M.; van Veldhuisen, D.J.; Muscholl, M.; Bergh, J.; Carlomagno, C.; Perren, T.; Passalacqua, R.; Bighin, C.; Klijn, J.G.; et al. Trastuzumab-associated cardiac adverse effects in the herceptin adjuvant trial. J. Clin. Oncol. 2007, 25, 3859–3865. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.M.; Wolf, M.J.; Rockman, H.A. Gene deletion screen for cardiomyopathy in adult Drosophila identifies a new notch ligand. Circ. Res. 2010, 106, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Neely, G.G.; Kuba, K.; Cammarato, A.; Isobe, K.; Amann, S.; Zhang, L.; Murata, M.; Elmen, L.; Gupta, V.; Arora, S.; et al. A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell 2010, 141, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tome-Esteban, M.T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Ware, S.M.; Quinn, M.E.; Ballard, E.T.; Miller, E.; Uzark, K.; Spicer, R.L. Pediatric restrictive cardiomyopathy associated with a mutation in beta-myosin heavy chain. Clin. Genet. 2008, 73, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Pasotti, M.; Pilotto, A.; Pellegrini, C.; Grasso, M.; Previtali, S.; Repetto, A.; Bellini, O.; Azan, G.; Scaffino, M.; et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur. J. Heart Fail. 2006, 8, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, F.; Lu, Q.W.; Morimoto, S.; Tanaka, H.; Kono, N.; Nagata, K.; Ojima, T.; Takahashi-Yanaga, F.; Miwa, Y.; Sasaguri, T.; et al. Drastic Ca2+ sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochem. Biophys. Res. Commun. 2005, 338, 1519–1526. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Balke, C.W.; Shorofsky, S.R. Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc. Res. 1998, 37, 290–299. [Google Scholar] [CrossRef]
- Kass, D.A.; Saavedra, W.F.; Sabbah, H.N. Reverse remodeling and enhanced inotropic reserve from the cardiac support device in experimental cardiac failure. J. Card. Fail. 2004, 10 (Suppl. 6), S215–S219. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S.; Jennings, T.; Dowse, H.; Ramaswami, M. Conditional mutations in SERCA, the Sarco-endoplasmic reticulum Ca2+-ATPase, alter heart rate and rhythmicity in Drosophila. J. Comp. Physiol. B 2005, 176, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.M.; Wolf, M.J. Disruption of Sarcoendoplasmic Reticulum Calcium ATPase Function in Drosophila Leads to Cardiac Dysfunction. PLoS ONE 2013, 8, e77785. [Google Scholar] [CrossRef] [PubMed]
- Magny, E.G.; Pueyo, J.I.; Pearl, F.M.G.; Cespedes, M.A.; Niven, J.E.; Bishop, S.A.; Couso, J.P. Conserved Regulation of Cardiac Calcium Uptake by Peptides Encoded in Small Open Reading Frames. Science 2013, 341, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.; Badie, N.; Yu, L.; Abraham, D.; Cheng, H.; Bursac, N.; Rockman, H.A.; Wolf, M.J. A Method to Measure Myocardial Calcium Handling in Adult Drosophila. Circ. Res. 2011, 108, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.M.C.; Scott, K.; Zuker, C.S.; Rubin, G.M. The ryanodine receptor is essential for larval development in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2000, 97, 5942–5947. [Google Scholar] [CrossRef] [PubMed]
- Ingwall, J.S.; Weiss, R.G. Is the Failing Heart Energy Starved? On Using Chemical Energy to Support Cardiac Function. Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Frenneaux, M.P.; Opie, L.H. Metabolic Mechanisms in Heart Failure. Circulation 2007, 116, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Diagnosis and Management of the Metabolic Syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef] [PubMed]
- Birse, R.T.; Choi, J.; Reardon, K.; Rodriguez, J.; Graham, S.; Diop, S.; Ocorr, K.; Bodmer, R.; Oldham, S. High-fat-diet-induced obesity and heart dysfunction are regulated by the TOR pathway in Drosophila. Cell Metab. 2010, 12, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Birse, R.T.; Bodmer, R. Lipotoxicity and cardiac dysfunction in mammals and Drosophila. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Diop, S.B.; Bodmer, R. Drosophila as a model to study the genetic mechanisms of obesity-associated heart dysfunction. J. Cell. Mol. Med. 2012, 16, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Na, J.; Musselman, L.P.; Pendse, J.; Baranski, T.J.; Bodmer, R.; Ocorr, K.; Cagan, R. A Drosophila model of high sugar diet-induced cardiomyopathy. PLoS Genet. 2013, 9, e1003175. [Google Scholar] [CrossRef] [PubMed]
- Diop, S.B.; Bisharat-Kernizan, J.; Birse, R.T.; Oldham, S.; Ocorr, K.; Bodmer, R. PGC-1/Spargel Counteracts High-Fat-Diet-Induced Obesity and Cardiac Lipotoxicity Downstream of TOR and Brummer ATGL Lipase. Cell Rep. 2015, 10, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Kelly, T.; He, J. Genetic epidemiology of obesity. Epidemiol. Rev. 2007, 29, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Le, H.D.; Melkani, G.C.; Panda, S. Time-restricted feeding attenuates age-related cardiac decline in Drosophila. Science 2015, 347, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Hatori, M.; Vollmers, C.; Zarrinpar, A.; DiTacchio, L.; Bushong, E.A.; Gill, S.; Leblanc, M.; Chaix, A.; Joens, M.; Fitzpatrick, J.A.J.; et al. Time-Restricted Feeding without Reducing Caloric Intake Prevents Metabolic Diseases in Mice Fed a High-Fat Diet. Cell Metab. 2012, 15, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Wessells, R.J.; Fitzgerald, E.; Cypser, J.R.; Tatar, M.; Bodmer, R. Insulin regulation of heart function in aging fruit flies. Nat. Genet. 2004, 36, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Wessells, R.; Fitzgerald, E.; Piazza, N.; Ocorr, K.; Morley, S.; Davies, C.; Lim, H.-Y.; Elmén, L.; Hayes, M.; Oldham, S.; et al. d4eBP acts downstream of both dTOR and dFoxo to modulate cardiac functional aging in Drosophila. Aging Cell 2009, 8, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Diop, S.B.; Bodmer, R. Gaining Insights into Diabetic Cardiomyopathy from Drosophila. Trends Endocrinol. Metab. 2015, 26, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Cannon, L.; Bodmer, R. Genetic manipulation of cardiac aging. J. Physiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.-Y.; Wang, W.; Chen, J.; Ocorr, K.; Bodmer, R. ROS regulate cardiac function via a distinct paracrine mechanism. Cell Rep. 2014, 7, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Melkani, G.C.; Trujillo, A.S.; Ramos, R.; Bodmer, R.; Bernstein, S.I.; Ocorr, K. Huntington’s disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart. PLoS Genet. 2013, 9, e1004024. [Google Scholar] [CrossRef] [PubMed]
- Monnier, V.; Iché-Torres, M.; Rera, M.; Contremoulins, V.; Guichard, C.; Lalevée, N.; Tricoire, H.; Perrin, L. dJun and Vri/dNFIL3 are major regulators of cardiac aging in Drosophila. PLoS Genet. 2012, 8, e1003081. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.B.; Cammarato, A.; Rajasekaran, N.S.; Zhang, H.; Suggs, J.A.; Lin, H.C.; Bernstein, S.I.; Benjamin, I.J.; Golic, K.G. The NADPH metabolic network regulates human alphaB-crystallin cardiomyopathy and reductive stress in Drosophila melanogaster. PLoS Genet. 2013, 9, e1003544. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, I.; Jablonska, J.; Zmojdzian, M.; Taghli-Lamallem, O.; Renaud, Y.; Junion, G.; Daczewska, M.; Huelsmann, S.; Jagla, K.; Jagla, T. Drosophila small heat shock protein CryAB ensures structural integrity of developing muscles, and proper muscle and heart performance. Development 2015, 142, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Fomovsky, G.M.; Thomopoulos, S.; Holmes, J.W. Contribution of extracellular matrix to the mechanical properties of the heart. J. Mol. Cell. Cardiol. 2010, 48, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Grossman, T.R.; Gamliel, A.; Wessells, R.J.; Taghli-Lamallem, O.; Jepsen, K.; Ocorr, K.; Korenberg, J.R.; Peterson, K.L.; Rosenfeld, M.G.; Bodmer, R.; et al. Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS Genet. 2011, 7, e1002344. [Google Scholar] [CrossRef] [PubMed]
- Dulcis, D.; Levine, R.B. Glutamatergic Innervation of the Heart Initiates Retrograde Contractions in Adult Drosophila melanogaster. J. Neurosci. 2005, 25, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- Lai, S.L.; Lee, T. Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat. Neurosci. 2006, 9, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.; Peabody, N.C.; Vinson, C.R.; White, B.H. Refined spatial manipulation of neuronal function by combinatorial restriction of transgene expression. Neuron 2006, 52, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.Y.; Gu, S.; Guttikonda, S.; Lin, T.Y.; White, B.H.; Lee, C.H. Focusing transgene expression in Drosophila by coupling Gal4 with a novel split-LexA expression system. Genetics 2011, 188, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Tasic, B.; Russler, E.V.; Liang, L.; Luo, L. The Q system: A repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell 2010, 141, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, L.; Taylor, M.R. Hearing the noise the challenges of human genome variation in genetic testing. J. Am. Coll. Cardiol. 2011, 57, 2328–2329. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Ackerman, M.J. The Achilles’ heel of cardiovascular genetic testing: Distinguishing pathogenic mutations from background genetic noise. Clin. Pharmacol. Ther. 2011, 90, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Marcou, C.A.; Tester, D.J. Personalized medicine: Genetic diagnosis for inherited cardiomyopathies/channelopathies. Rev. Esp. Cardiol. 2013, 66, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Hoogstra-Berends, F.; Meijering, R.A.M.; Zhang, D.; Heeres, A.; Loen, L.; Seerden, J.-P.; Kuipers, I.; Kampinga, H.H.; Henning, R.H.; Brundel, B.J.J.M. Heat Shock Protein-Inducing Compounds as Therapeutics to Restore Proteostasis in Atrial Fibrillation. Trends Cardiovasc. Med. 2012, 22, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, C.-T.; Qi, X.; Meijering, R.A.M.; Hoogstra-Berends, F.; Tadevosyan, A.; Cubukcuoglu-Deniz, G.; Durdu, S.; Akar, A.R.; Sibon, O.C.M.; et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of α-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Warf, M.B.; Nakamori, M.; Matthys, C.M.; Thornton, C.A.; Berglund, J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 18551–18556. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Selma-Soriano, E.; Magny, E.; Couso, J.P.; Pérez-Alonso, M.; Charlet-Berguerand, N.; Artero, R.; Llamusi, B. Pentamidine rescues contractility and rhythmicity in a Drosophila model of myotonic dystrophy heart dysfunction. Dis. Model. Mech. 2015, 8, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Tricoire, H.; Palandri, A.; Bourdais, A.; Camadro, J.-M.; Monnier, V. Methylene blue rescues heart defects in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Heidari, R.; Monnier, V.; Martin, E.; Tricoire, H. Methylene Blue Partially Rescues Heart Defects in a Drosophila Model of Huntington’s Disease. J. Huntingt. Dis. 2015, 4, 173–186. [Google Scholar] [CrossRef] [PubMed]



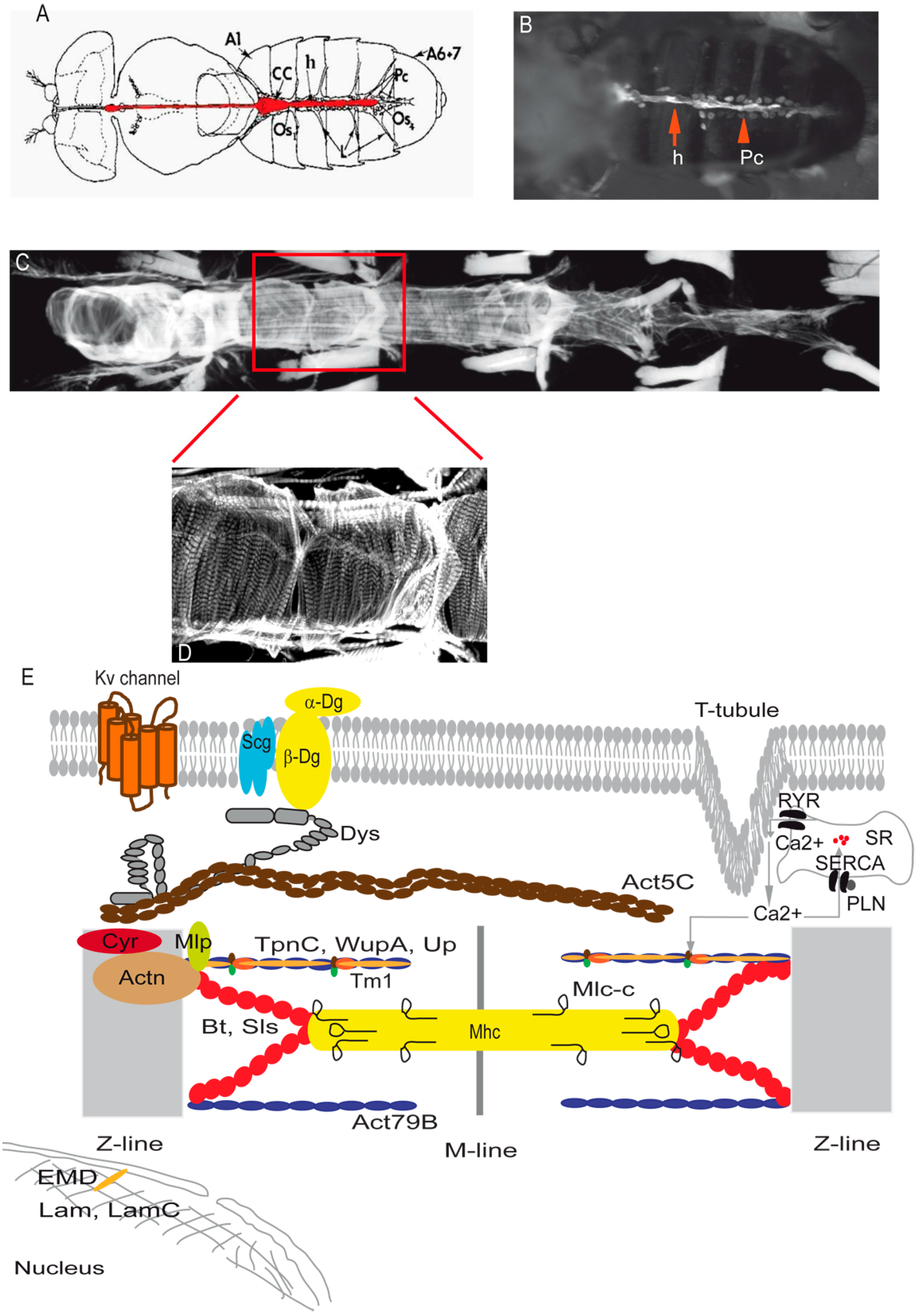{kind=link}
{kind=link}
| Disease Type | Human Gene | Drosophila Model | Major Findings in the Drosophila Model | References | |
|---|---|---|---|---|---|
| Channelopathies | K+ channels | KCNQ1 | KCNQ mutants | Increased arrhythmia and prolonged contractions | [48] |
| KCNK | ORK1 mutants | Increased HR | [40] | ||
| SUR2 | dSUR mutants | Heart failure Protective role against hypoxia and pacing-stress | [53] | ||
| KV2.1 | Shab mutants | Reduced HR | [54] | ||
| Cardiomyopathies | Hypertrophic CM | EGFR, RAS, RAF1 | Cardiac-specific expression of EGFR, Ras, Raf or Yki | Increased heart wall thickness Decreased cardiac lumen volume | [57,59] |
| Calcineurin | Cardiac-specific expression of constitutively activated CanA | Enlarged DD and reduced FS Heart wall thickening Identification of Galk as a repressor of CanA induced HCM | [60] | ||
| Dilated CM | TNNI3 and TPM1 | wupA and Tm1 mutants | Enlarged diameters and impaired systolic function | [50] | |
| DMD | Dys mutants | Dilated DD, SD and reduced FS DCM rescue by expressing mammalian form of Dys (Dp116) | [56] | ||
| SGCG | Human δ-ScgS151A/Large deletion of Scgδ | Enlarged heart tube, impaired systolic function and reduced FS | [50,122] | ||
| Myosin Heavy Chain (MYH7) | Hypoactive D45 Mhc mutants | Dilated heart and decreased FS | [55] | ||
| RHBDL2 | rho-3 mutants | Enlarged cardiac chamber rescued by expression of rho-3, spitz and EGFR | [123] | ||
| N/A | Cardiac-specific Wry RNAi and mutant | Enlarged DD/SD and decreased FS in wry mutants rescued by Notch expression Identification of Wry as a Notch ligand | [125] | ||
| CCR4-NOT3 | Cardiac-specific knockdown of Not3 | Increased DD, SD and reduced FS | [126] | ||
| Restrictive CM | Myosin Heavy Chain (MYH7) | Hyperactive Mhc5 mutants | Impaired diastolic function and restrictive heart phenotypes | [55] | |
| TNNT2 | Up101 mutants | Reduced DD, SD and FS Prolonged periods of systole and reduced HP | [58] | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taghli-Lamallem, O.; Plantié, E.; Jagla, K. Drosophila in the Heart of Understanding Cardiac Diseases: Modeling Channelopathies and Cardiomyopathies in the Fruitfly. J. Cardiovasc. Dev. Dis. 2016, 3, 7. https://doi.org/10.3390/jcdd3010007
Taghli-Lamallem O, Plantié E, Jagla K. Drosophila in the Heart of Understanding Cardiac Diseases: Modeling Channelopathies and Cardiomyopathies in the Fruitfly. Journal of Cardiovascular Development and Disease. 2016; 3(1):7. https://doi.org/10.3390/jcdd3010007
Chicago/Turabian StyleTaghli-Lamallem, Ouarda, Emilie Plantié, and Krzysztof Jagla. 2016. "Drosophila in the Heart of Understanding Cardiac Diseases: Modeling Channelopathies and Cardiomyopathies in the Fruitfly" Journal of Cardiovascular Development and Disease 3, no. 1: 7. https://doi.org/10.3390/jcdd3010007