Epac Function and cAMP Scaffolds in the Heart and Lung



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
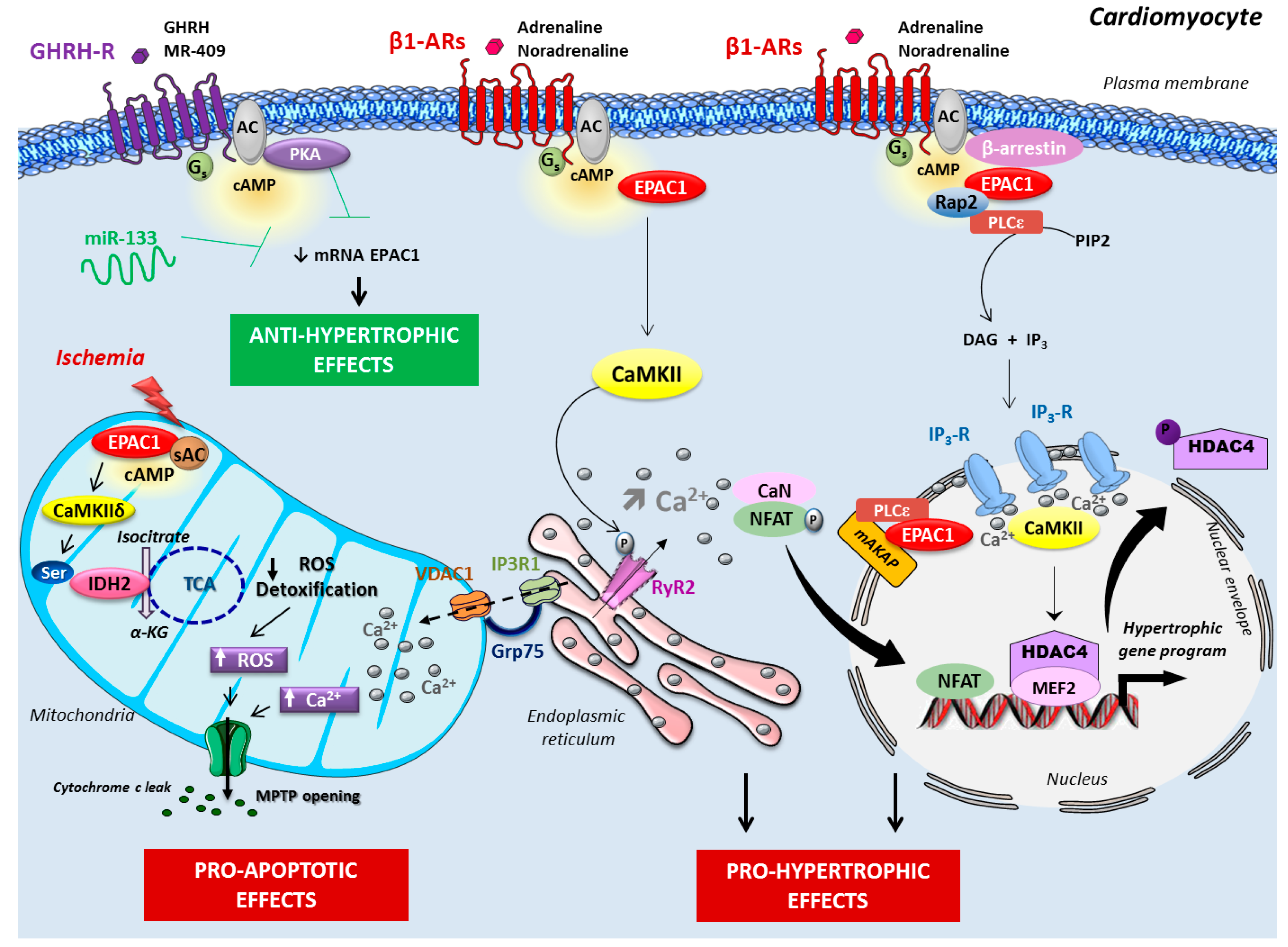
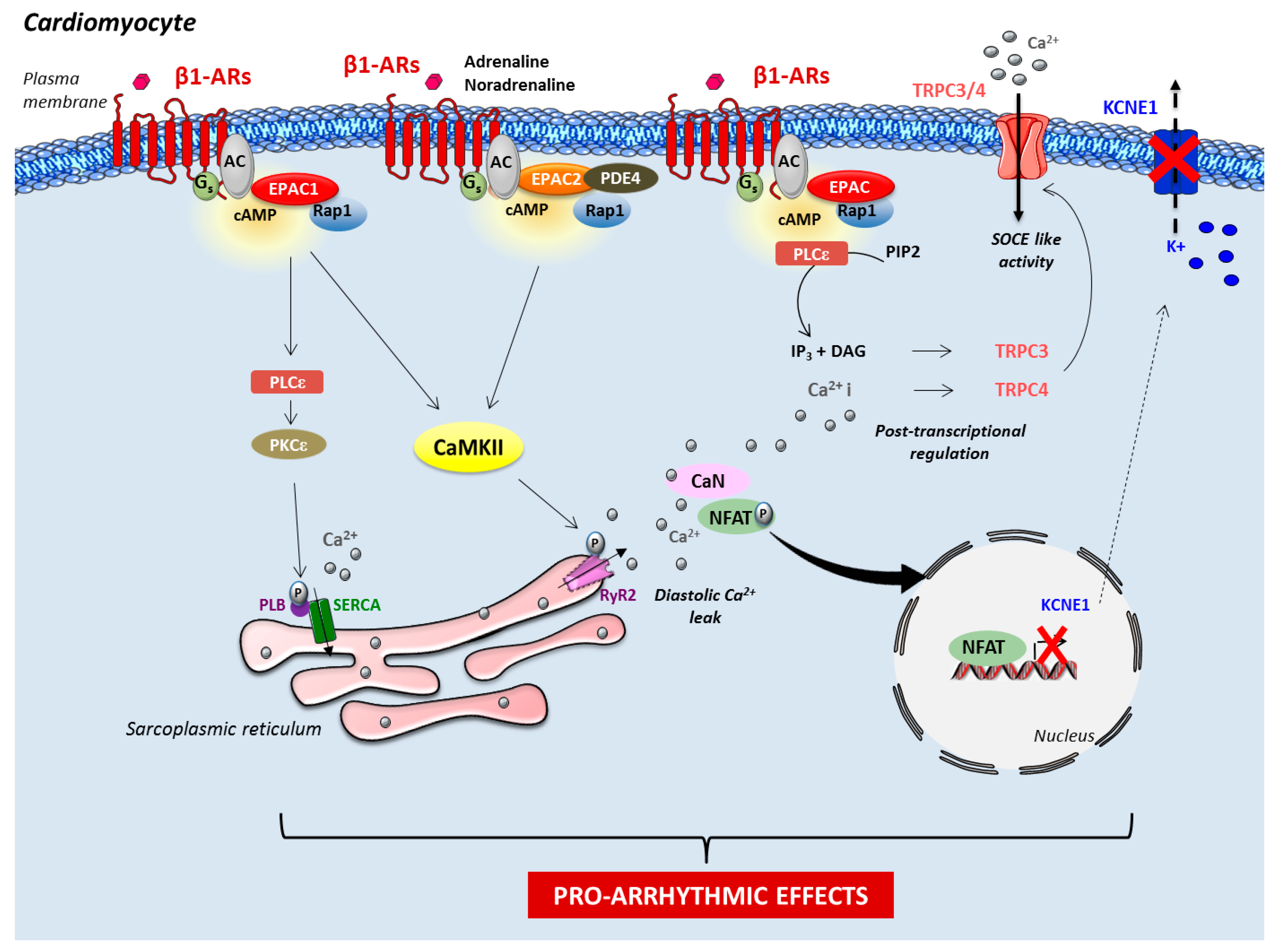
2. Epac in Cardiac Disease
2.1. Epac Signalosome in Pathological Cardiac Remodeling
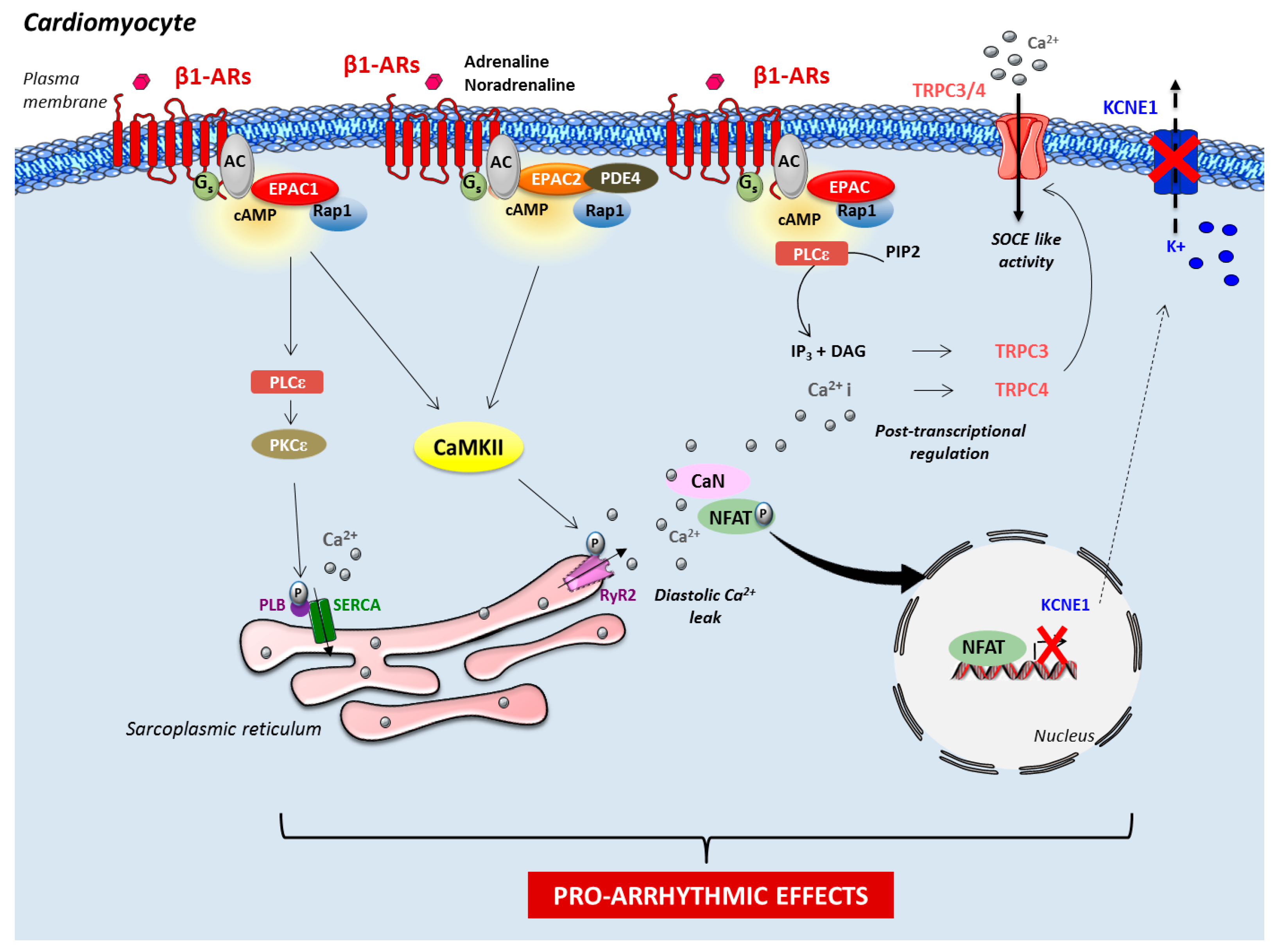
2.2. Role of Epac in Heart Failure and Arrhythmia
2.3. Role of Mitochondrial Epac in Cardiac Ischemia
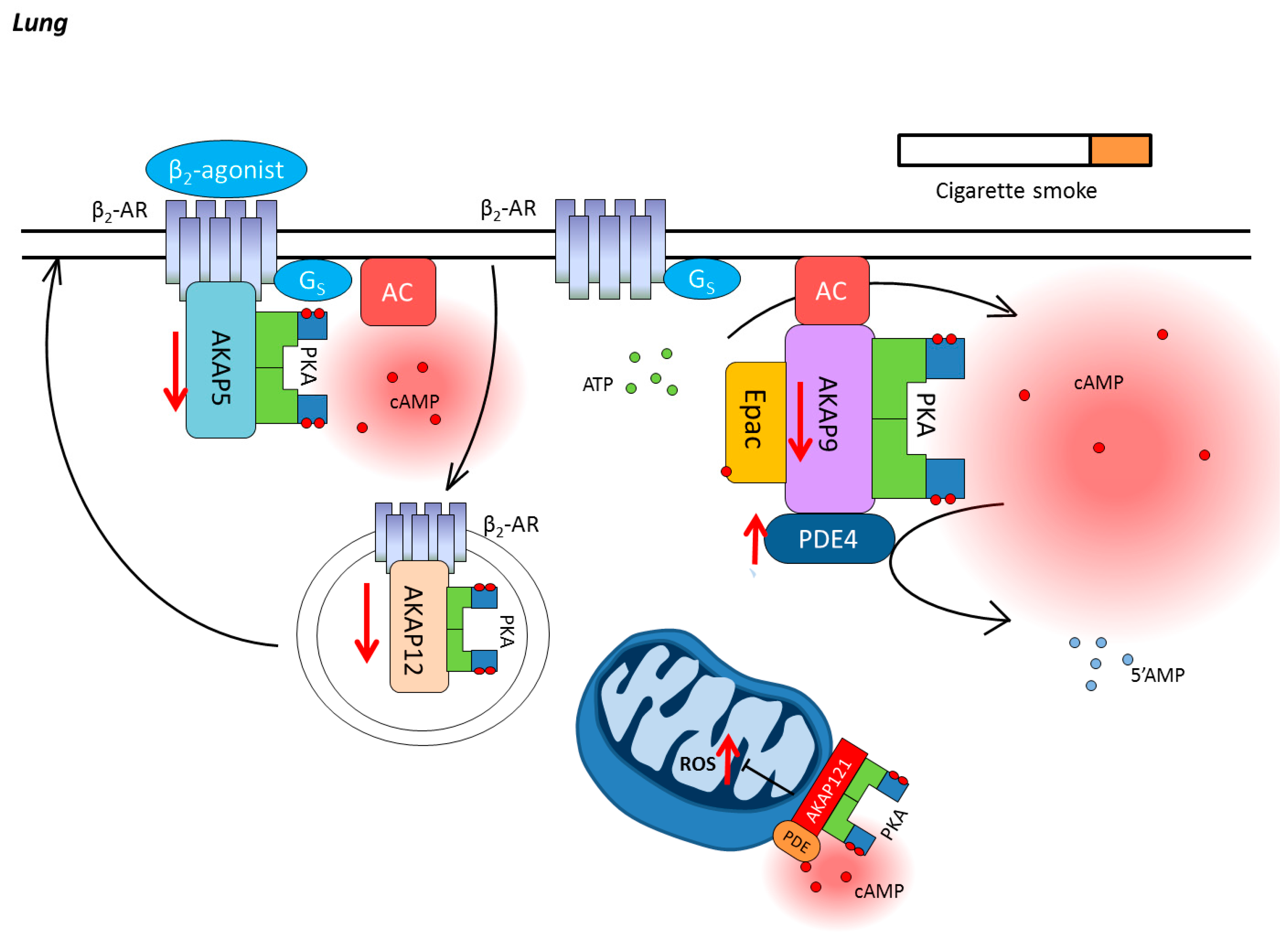
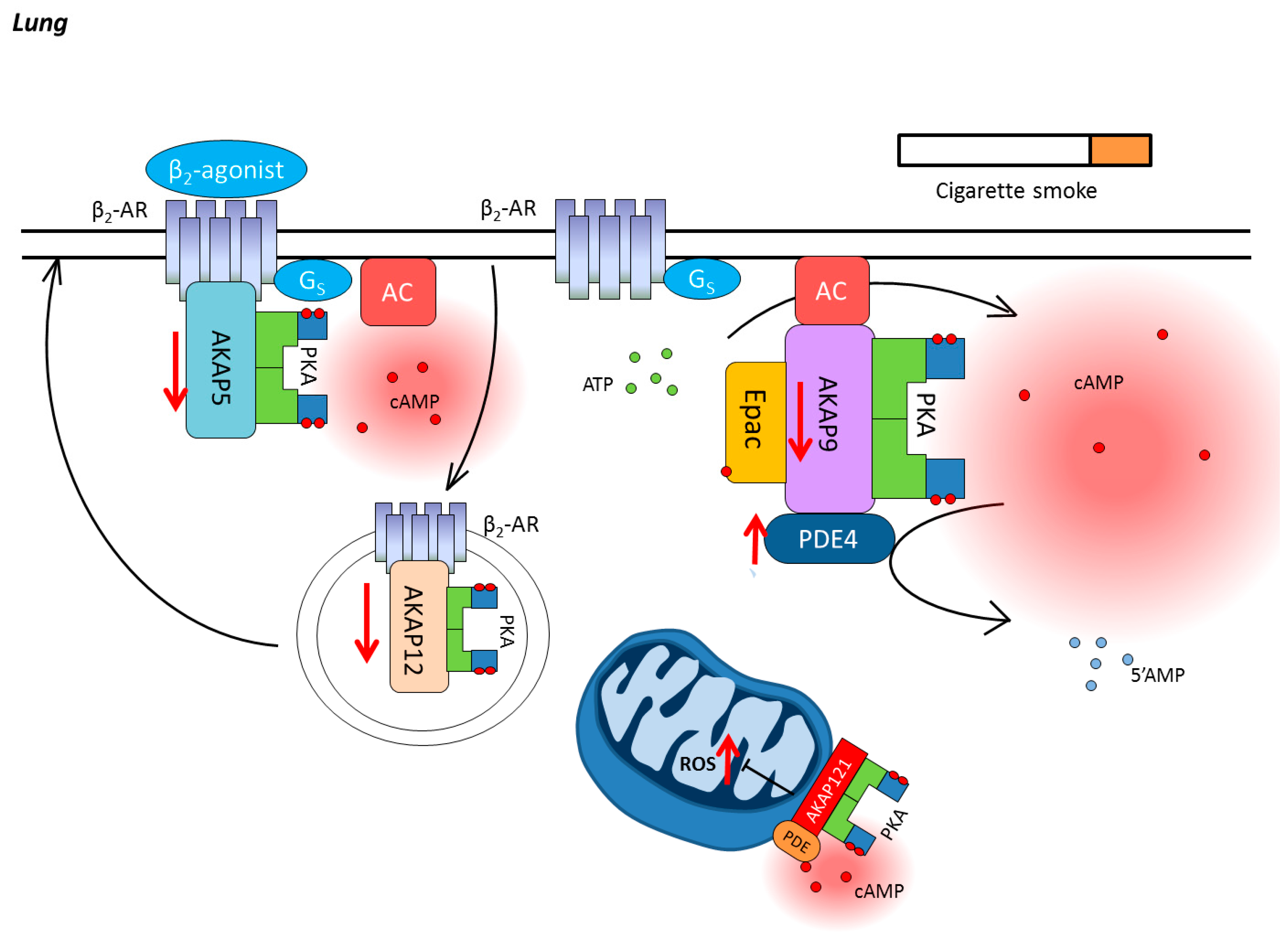
3. Chronic Obstructive Pulmonary Disease
3.1. Compartmentalization of cAMP in the Lung
3.2. Cyclic Nucleotide Phosphodiesterases in COPD
3.3. AKAPs in COPD
3.4. Epac in COPD
4. Conclusions and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Berthouze, M.; Laurent, A.-C.; Breckler, M.; Lezoualc’h, F. New perspectives in cAMP-signaling modulation. Curr. Heart Fail. Rep. 2011, 8, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R. Treatment of chronic heart failure with β-adrenergic receptor antagonists: A convergence of receptor pharmacology and clinical cardiology. Circ. Res. 2011, 109, 1176–1194. [Google Scholar] [CrossRef] [PubMed]
- El-Armouche, A.; Eschenhagen, T. β-adrenergic stimulation and myocardial function in the failing heart. Heart Fail. Rev. 2009, 14, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Von Lueder, T.G.; Krum, H. New medical therapies for heart failure. Nat. Rev. Cardiol. 2015, 12, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Levine, T.B.; Olivari, M.T.; Garberg, V.; Lura, D.; Francis, G.S.; Simon, A.B.; Rector, T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N. Engl. J. Med. 1984, 311, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Métrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.-L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac mediates β-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Ulucan, C.; Wang, X.; Baljinnyam, E.; Bai, Y.; Okumura, S.; Sato, M.; Minamisawa, S.; Hirotani, S.; Ishikawa, Y. Developmental changes in gene expression of Epac and its upregulation in myocardial hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1662–H1672. [Google Scholar] [CrossRef] [PubMed]
- Castaldi, A.; Zaglia, T.; Di Mauro, V.; Carullo, P.; Viggiani, G.; Borile, G.; Di Stefano, B.; Schiattarella, G.G.; Gualazzi, M.G.; Elia, L.; et al. MicroRNA-133 modulates the β1-adrenergic receptor transduction cascade. Circ. Res. 2014, 115, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Gesmundo, I.; Miragoli, M.; Carullo, P.; Trovato, L.; Larcher, V.; Di Pasquale, E.; Brancaccio, M.; Mazzola, M.; Villanova, T.; Sorge, M.; et al. Growth hormone-releasing hormone attenuates cardiac hypertrophy and improves heart function in pressure overload-induced heart failure. Proc. Natl. Acad. Sci. USA 2017, 114, 12033–12038. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Marcantoni, A.; Gastineau, M.; Birkedal, R.; Rochais, F.; Garnier, A.; Lompré, A.-M.; Vandecasteele, G.; Lezoualc’h, F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res. 2005, 97, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Schwede, F.; Bertinetti, D.; Langerijs, C.N.; Hadders, M.A.; Wienk, H.; Ellenbroek, J.H.; de Koning, E.J.P.; Bos, J.L.; Herberg, F.W.; Genieser, H.-G.; et al. Structure-guided design of selective Epac1 and Epac2 agonists. PLoS Biol. 2015, 13, e1002038. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.-C.; Bisserier, M.; Lucas, A.; Tortosa, F.; Roumieux, M.; De Régibus, A.; Swiader, A.; Sainte-Marie, Y.; Heymes, C.; Vindis, C.; et al. Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc. Res. 2015, 105, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Blondeau, J.-P.; Lezoualc’h, F. Epac proteins: Specific ligands and role in cardiac remodelling. Biochem. Soc. Trans. 2014, 42, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Courilleau, D.; Bisserier, M.; Jullian, J.-C.; Lucas, A.; Bouyssou, P.; Fischmeister, R.; Blondeau, J.-P.; Lezoualc’h, F. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J. Biol. Chem. 2012, 287, 44192–44202. [Google Scholar] [CrossRef] [PubMed]
- Monceau, V.; Llach, A.; Azria, D.; Bridier, A.; Petit, B.; Mazevet, M.; Strup-Perrot, C.; To, T.-H.-V.; Calmels, L.; Germaini, M.-M.; et al. Epac contributes to cardiac hypertrophy and amyloidosis induced by radiotherapy but not fibrosis. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2014, 111, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, O.; Lucas, A.; Poirier, F.; Lacampagne, A.; Lezoualc’h, F. The cAMP binding protein Epac regulates cardiac myofilament function. Proc. Natl. Acad. Sci. USA 2009, 106, 14144–14149. [Google Scholar] [CrossRef] [PubMed]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Rehmann, H.; Lao, D.H.; Erickson, J.R.; Bossuyt, J.; Chen, J.; Bers, D.M. Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 3991–3996. [Google Scholar] [CrossRef] [PubMed]
- Mangmool, S.; Shukla, A.K.; Rockman, H.A. β-Arrestin-dependent activation of Ca2+/calmodulin kinase II after β1-adrenergic receptor stimulation. J. Cell Biol. 2010, 189, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Berthouze-Duquesnes, M.; Lucas, A.; Saulière, A.; Sin, Y.Y.; Laurent, A.-C.; Galés, C.; Baillie, G.; Lezoualc’h, F. Specific interactions between Epac1, β-arrestin2 and PDE4D5 regulate β-adrenergic receptor subtype differential effects on cardiac hypertrophic signaling. Cell. Signal. 2013, 25, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.L.; Soughayer, J.; Pare, G.C.; Carlisle Michel, J.J.; Langeberg, L.K.; Kapiloff, M.S.; Scott, J.D. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature 2005, 437, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Malik, S.; Pang, J.; Wang, H.; Park, K.M.; Yule, D.I.; Blaxall, B.C.; Smrcka, A.V. Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 2013, 153, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Ruiz-Hurtado, G.; Morel, E.; Laurent, A.-C.; Métrich, M.; Domínguez-Rodríguez, A.; Lauton-Santos, S.; Lucas, A.; Benitah, J.-P.; Bers, D.M.; et al. Epac enhances excitation-transcription coupling in cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Métrich, M.; Laurent, A.-C.; Breckler, M.; Duquesnes, N.; Hmitou, I.; Courillau, D.; Blondeau, J.-P.; Crozatier, B.; Lezoualc’h, F.; Morel, E. Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell. Signal. 2010, 22, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Hurtado, G.; Morel, E.; Domínguez-Rodríguez, A.; Llach, A.; Lezoualc’h, F.; Benitah, J.-P.; Gomez, A.M. Epac in cardiac calcium signaling. J. Mol. Cell. Cardiol. 2013, 58, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Fujita, T.; Cai, W.; Jin, M.; Namekata, I.; Mototani, Y.; Jin, H.; Ohnuki, Y.; Tsuneoka, Y.; Kurotani, R.; et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J. Clin. Investig. 2014, 124, 2785–2801. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Cheng, H.; Lao, D.H.; Na, L.; van Oort, R.J.; Brown, J.H.; Wehrens, X.H.T.; Chen, J.; Bers, D.M. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 2013, 127, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Kong, C.H.T.; Cannell, M.B.; Ward, M.-L. Depotentiation of intact rat cardiac muscle unmasks an Epac-dependent increase in myofilament Ca2+ sensitivity. Clin. Exp. Pharmacol. Physiol. 2016, 43, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Lezcano, N.; Mariángelo, J.I.E.; Vittone, L.; Wehrens, X.H.T.; Said, M.; Mundiña-Weilenmann, C. Early effects of Epac depend on the fine-tuning of the sarcoplasmic reticulum Ca2+ handling in cardiomyocytes. J. Mol. Cell. Cardiol. 2017, 114, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, E.A.; Wang, H.; Malik, S.; Kaproth-Joslin, K.A.; Blaxall, B.C.; Kelley, G.G.; Dirksen, R.T.; Smrcka, A.V. Epac-mediated activation of phospholipase C(epsilon) plays a critical role in β-adrenergic receptor-dependent enhancement of Ca2+ mobilization in cardiac myocytes. J. Biol. Chem. 2007, 282, 5488–5495. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Bare, D.J.; Galice, S.; Shannon, T.R.; Bers, D.M. β-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J. Mol. Cell. Cardiol. 2017, 108, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Bobin, P.; Varin, A.; Lefebvre, F.; Fischmeister, R.; Vandecasteele, G.; Leroy, J. Calmodulin kinase II inhibition limits the pro-arrhythmic Ca2+ waves induced by cAMP-phosphodiesterase inhibitors. Cardiovasc. Res. 2016, 110, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Fujita, T.; Hidaka, Y.; Jin, H.; Suita, K.; Prajapati, R.; Liang, C.; Umemura, M.; Yokoyama, U.; Sato, M.; et al. Disruption of Epac1 protects the heart from adenylyl cyclase type 5-mediated cardiac dysfunction. Biochem. Biophys. Res. Commun. 2016, 475, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hothi, S.S.; Gurung, I.S.; Heathcote, J.C.; Zhang, Y.; Booth, S.W.; Skepper, J.N.; Grace, A.A.; Huang, C.L.-H. Epac activation, altered calcium homeostasis and ventricular arrhythmogenesis in the murine heart. Pflug. Arch. 2008, 457, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Hurtado, G.; Domínguez-Rodríguez, A.; Pereira, L.; Fernández-Velasco, M.; Cassan, C.; Lezoualc’h, F.; Benitah, J.-P.; Gómez, A.M. Sustained Epac activation induces calmodulin dependent positive inotropic effect in adult cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 53, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Brette, F.; Blandin, E.; Simard, C.; Guinamard, R.; Sallé, L. Epac activator critically regulates action potential duration by decreasing potassium current in rat adult ventricle. J. Mol. Cell. Cardiol. 2013, 57, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, M.; Qi, X.-Y.; Xiao, L.; Ordog, B.; Tadevosyan, A.; Luo, X.; Maguy, A.; Shi, Y.; Tardif, J.-C.; Nattel, S. Exchange protein directly activated by cAMP mediates slow delayed-rectifier current remodeling by sustained β-adrenergic activation in guinea pig hearts. Circ. Res. 2014, 114, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Rodríguez, A.; Ruiz-Hurtado, G.; Sabourin, J.; Gómez, A.M.; Alvarez, J.L.; Benitah, J.-P. Proarrhythmic effect of sustained EPAC activation on TRPC3/4 in rat ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2015, 87, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Duquesnes, N.; Derangeon, M.; Métrich, M.; Lucas, A.; Mateo, P.; Li, L.; Morel, E.; Lezoualc’h, F.; Crozatier, B. Epac stimulation induces rapid increases in connexin43 phosphorylation and function without preconditioning effect. Pflugers Arch. 2010, 460, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Somekawa, S.; Fukuhara, S.; Nakaoka, Y.; Fujita, H.; Saito, Y.; Mochizuki, N. Enhanced functional gap junction neoformation by protein kinase A-dependent and Epac-dependent signals downstream of cAMP in cardiac myocytes. Circ. Res. 2005, 97, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Métrich, M.; Fernández-Velasco, M.; Lucas, A.; Leroy, J.; Perrier, R.; Morel, E.; Fischmeister, R.; Richard, S.; Bénitah, J.-P.; et al. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. 2007, 583, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kirton, H.M.; Al-Owais, M.; Thireau, J.; Richard, S.; Peers, C.; Steele, D.S. Epac2-Rap1 Signaling Regulates Reactive Oxygen Species Production and Susceptibility to Cardiac Arrhythmias. Antioxid. Redox Signal. 2017, 27, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Treatment of Myocardial Ischemia/Reperfusion Injury by Ischemic and Pharmacological Postconditioning. Compr. Physiol. 2015, 5, 1123–1145. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Yokoyama, U.; Abe, T.; Kiyonari, H.; Yamashita, N.; Kato, Y.; Kurotani, R.; Sato, M.; Okumura, S.; Ishikawa, Y. Differential roles of Epac in regulating cell death in neuronal and myocardial cells. J. Biol. Chem. 2010, 285, 24248–24259. [Google Scholar] [CrossRef] [PubMed]
- Mangmool, S.; Hemplueksa, P.; Parichatikanond, W.; Chattipakorn, N. Epac is required for GLP-1R-mediated inhibition of oxidative stress and apoptosis in cardiomyocytes. Mol. Endocrinol. 2015, 29, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Khaliulin, I.; Bond, M.; James, A.F.; Dyar, Z.; Amini, R.; Johnson, J.L.; Suleiman, M.-S. Functional and cardioprotective effects of simultaneous and individual activation of protein kinase A and Epac. Br. J. Pharmacol. 2017, 174, 438–453. [Google Scholar] [CrossRef] [PubMed]
- Edland, F.; Wergeland, A.; Kopperud, R.; Åsrud, K.S.; Hoivik, E.A.; Witsø, S.L.; Æsøy, R.; Madsen, L.; Kristiansen, K.; Bakke, M.; et al. Long-term consumption of an obesogenic high fat diet prior to ischemia-reperfusion mediates cardioprotection via Epac1-dependent signaling. Nutr. Metab. 2016, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Sánchez, E.; Díaz, I.; Ordóñez, A.; Smani, T. Urocortin-1 Mediated Cardioprotection Involves XIAP and CD40-Ligand Recovery: Role of EPAC2 and ERK1/2. PLoS ONE 2016, 11, e0147375. [Google Scholar] [CrossRef] [PubMed]
- Fazal, L.; Laudette, M.; Paula-Gomes, S.; Pons, S.; Conte, C.; Tortosa, F.; Sicard, P.; Sainte-Marie, Y.; Bisserier, M.; Lairez, O.; et al. Multifunctional Mitochondrial Epac1 Controls Myocardial Cell Death. Circ. Res. 2017, 120, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Mei, F.C.; Popov, V.L.; Vergara, L.A.; Cheng, X. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J. Biol. Chem. 2002, 277, 26581–26586. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, D.; Varin, A.; Nicolas, V.; Courilleau, D.; Mateo, P.; Caubere, C.; Rouet, P.; Gomez, A.-M.; Vandecasteele, G.; et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death Dis. 2016, 7, e2198. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.-A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.who.int/respiratory/copd/en/ (accessed on 31 January 2018).
- Antus, B.; Kardos, Z. Oxidative stress in COPD: Molecular background and clinical monitoring. Curr. Med. Chem. 2015, 22, 627–650. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.A.; Barnes, P.J. Oxidative Stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, C.; Boulet, L.-P. Structural changes in airway diseases: Characteristics, mechanisms, consequences, and pharmacologic modulation. Chest 2006, 129, 1068–1087. [Google Scholar] [CrossRef] [PubMed]
- Tønnesen, P. Smoking cessation and COPD. Eur. Respir. Rev. 2013, 22, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Boswell-Smith, V.; Spina, D. PDE4 inhibitors as potential therapeutic agents in the treatment of COPD-focus on roflumilast. Int. J. Chronic Obstr. Pulm. Dis. 2007, 2, 121–129. [Google Scholar]
- Pauwels, R.A.; Buist, A.S.; Calverley, P.M.; Jenkins, C.R.; Hurd, S.S. GOLD Scientific Committee Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am. J. Respir. Crit. Care Med. 2001, 163, 1256–1276. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, B.G.J.; Racké, K.; Schmidt, M. Distinct PKA and Epac compartmentalization in airway function and plasticity. Pharmacol. Ther. 2013, 137, 248–265. [Google Scholar] [CrossRef] [PubMed]
- Kabir, E.R.; Morshed, N. Different approaches in the treatment of obstructive pulmonary diseases. Eur. J. Pharmacol. 2015, 764, 306–317. [Google Scholar] [CrossRef] [PubMed]
- López-Campos, J.L.; Calero Acuña, C. What is in the guidelines about the pharmacological treatment of chronic obstructive pulmonary disease? Expert Rev. Respir. Med. 2013, 7, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Qaseem, A.; Wilt, T.J.; Weinberger, S.E.; Hanania, N.A.; Criner, G.; van der Molen, T.; Marciniuk, D.D.; Denberg, T.; Schünemann, H.; Wedzicha, W.; et al. Diagnosis and management of stable chronic obstructive pulmonary disease: A clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann. Intern. Med. 2011, 155, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Abbott-Banner, K.H.; Page, C.P. Dual PDE3/4 and PDE4 inhibitors: Novel treatments for COPD and other inflammatory airway diseases. Basic Clin. Pharmacol. Toxicol. 2014, 114, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Raissy, H.H.; Kelly, H.W.; Harkins, M.; Szefler, S.J. Inhaled Corticosteroids in Lung Diseases. Am. J. Respir. Crit. Care Med. 2013, 187, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Dekker, F.J.; Maarsingh, H. Exchange Protein Directly Activated by cAMP (epac): A Multidomain cAMP Mediator in the Regulation of Diverse Biological Functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [PubMed]
- Hanoune, J.; Defer, N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.J.; Deshpande, D.A.; Tiegs, B.C.; Misior, A.M.; Yan, H.; Hershfeld, A.V.; Rich, T.C.; Panettieri, R.A.; An, S.S.; Penn, R.B. β-Agonist-mediated relaxation of airway smooth muscle is protein kinase A-dependent. J. Biol. Chem. 2014, 289, 23065–23074. [Google Scholar] [CrossRef] [PubMed]
- Roscioni, S.S.; Maarsingh, H.; Elzinga, C.R.S.; Schuur, J.; Menzen, M.; Halayko, A.J.; Meurs, H.; Schmidt, M. Epac as a novel effector of airway smooth muscle relaxation. J. Cell. Mol. Med. 2011, 15, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Maher, S.A.; Dekkak, B.; Jones, V.; Wong, S.; Brook, P.; Belvisi, M.G. Anti-inflammatory effects of PGE2 in the lung: Role of the EP4 receptor subtype. Thorax 2015, 70, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Sarriá, B.; Buenestado, A.; Cortijo, J.; Cerdá, M.; Morcillo, E.J. Phosphodiesterase 4 inhibition decreases MUC5AC expression induced by epidermal growth factor in human airway epithelial cells. Thorax 2005, 60, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Profita, M.; Chiappara, G.; Mirabella, F.; Di Giorgi, R.; Chimenti, L.; Costanzo, G.; Riccobono, L.; Bellia, V.; Bousquet, J.; Vignola, A.M. Effect of cilomilast (Ariflo) on TNF-α, IL-8, and GM-CSF release by airway cells of patients with COPD. Thorax 2003, 58, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.A.; Poole, J.A.; Nordgren, T.M.; DeVasure, J.M.; Heires, A.J.; Bailey, K.L.; Romberger, D.J. cAMP-dependent protein kinase activation decreases cytokine release in bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L643–L651. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Wettlaufer, S.H.; Hogaboam, C.; Aronoff, D.M.; Peters-Golden, M. Prostaglandin E2 inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L405–L413. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Means, C.K.; Scott, J.D. A-Kinase Anchoring Proteins: From protein complexes to physiology and disease. IUBMB Life 2009, 61, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Poppinga, W.J.; Muñoz-Llancao, P.; González-Billault, C.; Schmidt, M. A-kinase anchoring proteins: cAMP compartmentalization in neurodegenerative and obstructive pulmonary diseases. Br. J. Pharmacol. 2014, 171, 5603–5623. [Google Scholar] [CrossRef] [PubMed]
- Page, C.P.; Spina, D. Selective PDE inhibitors as novel treatments for respiratory diseases. Curr. Opin. Pharmacol. 2012, 12, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.; Baillie, G.S.; Bergmann, R.; Shepherd, M.C.; Sepper, R.; Houslay, M.D.; Heeke, G.V. Differential expression of PDE4 cAMP phosphodiesterase isoforms in inflammatory cells of smokers with COPD, smokers without COPD, and nonsmokers. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L332–L343. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.-K.; Hu, H.-J.; Rhee, C.-K.; Shin, S.-H.; Oh, Y.-M.; Lee, S.-D.; Jung, S.-H.; Yim, S.-H.; Kim, T.-M. Korean Obstructive Lung Disease (KOLD) Study Group; et al. Polymorphisms in PDE4D are associated with a risk of COPD in non-emphysematous Koreans. COPD 2014, 11, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Trian, T.; Burgess, J.K.; Niimi, K.; Moir, L.M.; Ge, Q.; Berger, P.; Liggett, S.B.; Black, J.L.; Oliver, B.G. β2-Agonist Induced cAMP Is Decreased in Asthmatic Airway Smooth Muscle Due to Increased PDE4D. PLoS ONE 2011, 6, e20000. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Lluch, J.; Almudever, P.; Freire, J.; Xiaozhong, Q.; Cortijo, J. Roflumilast N-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2014, 134, 314–322.e9. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Armengot, M.; Bañuls, P.; Tenor, H.; Beume, R.; Artigues, E.; Cortijo, J. Roflumilast N-oxide, a PDE4 inhibitor, improves cilia motility and ciliated human bronchial epithelial cells compromised by cigarette smoke in vitro. Br. J. Pharmacol. 2012, 166, 2243–2262. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.; Robas, N.; Cawkill, D.; Fidock, M. Cloning and characterization of the human and mouse PDE7B, a novel cAMP-specific cyclic nucleotide phosphodiesterase. Biochem. Biophys. Res. Commun. 2000, 272, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Hetman, J.M.; Soderling, S.H.; Glavas, N.A.; Beavo, J.A. Cloning and characterization of PDE7B, a cAMP-specific phosphodiesterase. Proc. Natl. Acad. Sci. USA 2000, 97, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, M.; Jahn, H.-U.; Seybold, J.; Neurohr, C.; Barnes, P.J.; Hippenstiel, S.; Kraemer, H.J.; Suttorp, N. Identification and Function of Cyclic Nucleotide Phosphodiesterase Isoenzymes in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 1999, 20, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Brookes-Fazakerley, S.; Donnelly, L.E.; Barnes, P.J.; Barnette, M.S.; Giembycz, M.A. Ubiquitous expression of phosphodiesterase 7A in human proinflammatory and immune cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L279–L289. [Google Scholar] [CrossRef] [PubMed]
- Giembycz, M.A.; Smith, S.J. Phosphodiesterase 7A: A new therapeutic target for alleviating chronic inflammation? Curr. Pharm. Des. 2006, 12, 3207–3220. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Cieslinski, L.B.; Newton, R.; Donnelly, L.E.; Fenwick, P.S.; Nicholson, A.G.; Barnes, P.J.; Barnette, M.S.; Giembycz, M.A. Discovery of BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene], a Selective Inhibitor of Phosphodiesterase 7: In Vitro Studies in Human Monocytes, Lung Macrophages, and CD8+ T-Lymphocytes. Mol. Pharmacol. 2004, 66, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Cong, M.; Perry, S.J.; Lin, F.T.; Fraser, I.D.; Hu, L.A.; Chen, W.; Pitcher, J.A.; Scott, J.D.; Lefkowitz, R.J. Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J. Biol. Chem. 2001, 276, 15192–15199. [Google Scholar] [CrossRef] [PubMed]
- Fraser, I.D.; Cong, M.; Kim, J.; Rollins, E.N.; Daaka, Y.; Lefkowitz, R.J.; Scott, J.D. Assembly of an A kinase-anchoring protein-β2-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr. Biol. 2000, 10, 409–412. [Google Scholar] [CrossRef]
- Tao, J.; Malbon, C.C. G-protein-coupled receptor-associated A-kinase anchoring proteins AKAP5 and AKAP12: Differential signaling to MAPK and GPCR recycling. J. Mol. Signal. 2008, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Poppinga, W.J.; Heijink, I.H.; Holtzer, L.J.; Skroblin, P.; Klussmann, E.; Halayko, A.J.; Timens, W.; Maarsingh, H.; Schmidt, M. A-kinase-anchoring proteins coordinate inflammatory responses to cigarette smoke in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L766–L775. [Google Scholar] [CrossRef] [PubMed]
- Oldenburger, A.; Poppinga, W.J.; Kos, F.; de Bruin, H.G.; Rijks, W.F.; Heijink, I.H.; Timens, W.; Meurs, H.; Maarsingh, H.; Schmidt, M. A-kinase anchoring proteins contribute to loss of E-cadherin and bronchial epithelial barrier by cigarette smoke. Am. J. Physiol. Cell Physiol. 2014, 306, C585–C597. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Kass, R.S.; Dessauer, C.W. The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J. Biol. Chem. 2012, 287, 29815–29824. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Feliciello, A.; Schiattarella, G.G.; Esposito, G.; Guerriero, R.; Zaccaro, L.; Del Gatto, A.; Saviano, M.; Garbi, C.; Carangi, R.; et al. AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc. Res. 2010, 88, 101–110. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.; Harris, G.J.; Pinder, E.M.; Macfarlane, J.G.; Hellyer, T.P.; Rostron, A.J.; Conway Morris, A.; Thickett, D.R.; Perkins, G.D.; McAuley, D.F.; et al. Exchange protein directly activated by cyclic AMP (EPAC) activation reverses neutrophil dysfunction induced by β2-agonists, corticosteroids, and critical illness. J. Allergy Clin. Immunol. 2016, 137, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Oldenburger, A.; Roscioni, S.S.; Jansen, E.; Menzen, M.H.; Halayko, A.J.; Timens, W.; Meurs, H.; Maarsingh, H.; Schmidt, M. Anti-inflammatory role of the cAMP effectors Epac and PKA: Implications in chronic obstructive pulmonary disease. PLoS ONE 2012, 7, e31574. [Google Scholar] [CrossRef] [PubMed]
- Grandoch, M.; Roscioni, S.S.; Schmidt, M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br. J. Pharmacol. 2010, 159, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Oldenburger, A.; van Basten, B.; Kooistra, W.; Meurs, H.; Maarsingh, H.; Krenning, G.; Timens, W.; Schmidt, M. Interaction between Epac1 and miRNA-7 in airway smooth muscle cells. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Oldenburger, A.; Timens, W.; Bos, S.; Smit, M.; Smrcka, A.V.; Laurent, A.-C.; Cao, J.; Hylkema, M.; Meurs, H.; Maarsingh, H.; et al. Epac1 and Epac2 are differentially involved in inflammatory and remodeling processes induced by cigarette smoke. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4617–4628. [Google Scholar] [CrossRef] [PubMed]
- Börner, S.; Schwede, F.; Schlipp, A.; Berisha, F.; Calebiro, D.; Lohse, M.J.; Nikolaev, V.O. FRET measurements of intracellular cAMP concentrations and cAMP analog permeability in intact cells. Nat. Protoc. 2011, 6, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Kinkorová, J. Biobanks in the era of personalized medicine: Objectives, challenges, and innovation. EPMA J. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laudette, M.; Zuo, H.; Lezoualc’h, F.; Schmidt, M. Epac Function and cAMP Scaffolds in the Heart and Lung. J. Cardiovasc. Dev. Dis. 2018, 5, 9. https://doi.org/10.3390/jcdd5010009
Laudette M, Zuo H, Lezoualc’h F, Schmidt M. Epac Function and cAMP Scaffolds in the Heart and Lung. Journal of Cardiovascular Development and Disease. 2018; 5(1):9. https://doi.org/10.3390/jcdd5010009
Chicago/Turabian StyleLaudette, Marion, Haoxiao Zuo, Frank Lezoualc’h, and Martina Schmidt. 2018. "Epac Function and cAMP Scaffolds in the Heart and Lung" Journal of Cardiovascular Development and Disease 5, no. 1: 9. https://doi.org/10.3390/jcdd5010009