TREM-1; Is It a Pivotal Target for Cardiovascular Diseases?


Abstract
:1. Introduction
2. TREM-1 Signaling
TREM-1 Ligands
3. TREM-1 in Inflammatory Processes
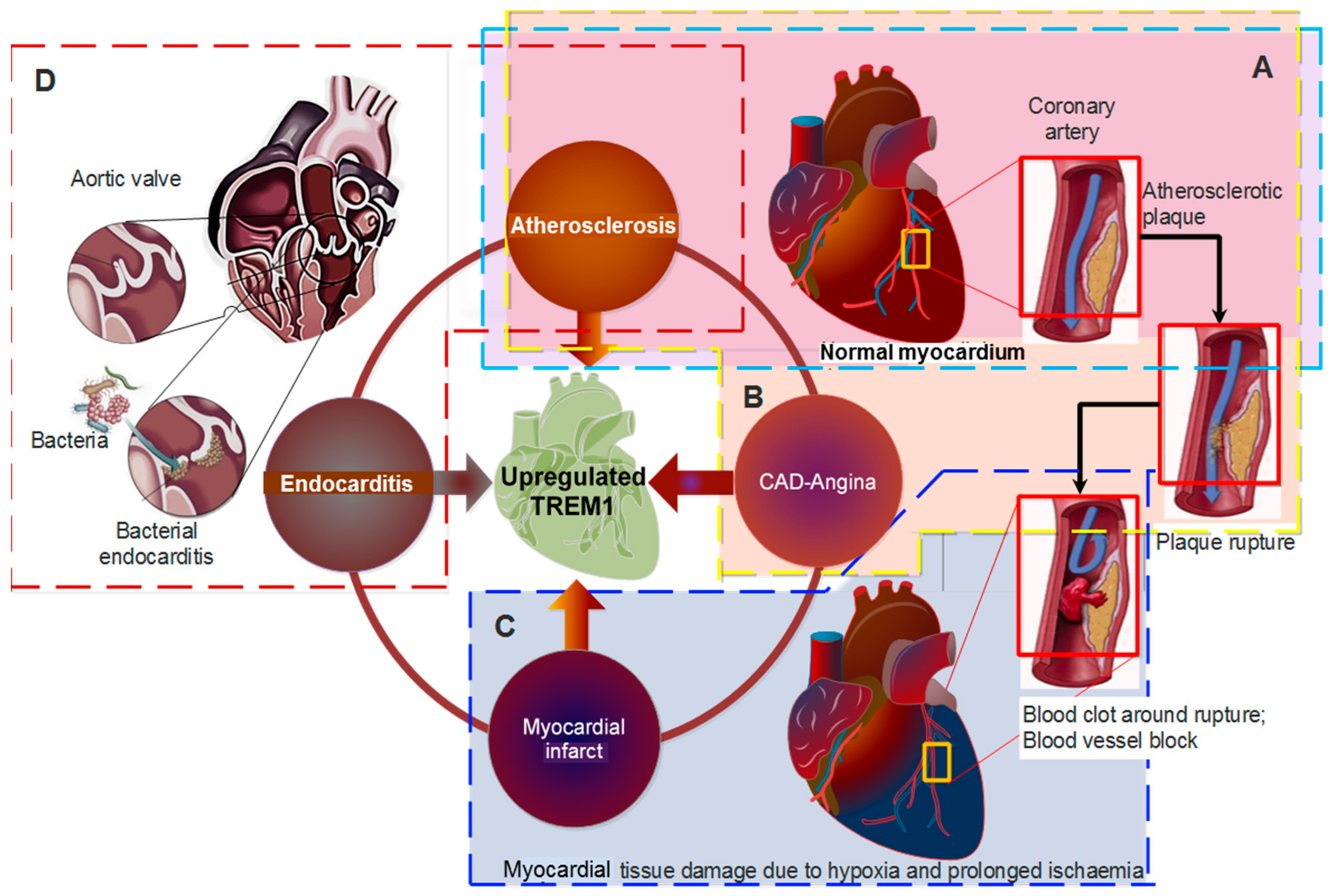
4. TREM-1 in Cardiovascular Diseases
4.1. TREM-1 in Pathogenesis of Atherosclerosis
4.2. TREM-1 in Coronary Artery Disease (CAD) and Acute Myocardial Infarction (AMI)
4.3. TREM-1 in Endocarditis
5. Therapeutics of Cardiovascular Diseases Targeting TREM1 Pathway
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sharma, A.; Green, J.B.; Dunning, A.; Lokhnygina, Y.; Al-Khatib, S.M.; Lopes, R.D.; Buse, J.B.; Lachin, J.M.; Van de Werf, F.; Armstrong, P.W.; et al. Causes of Death in a Contemporary Cohort of Patients with Type 2 Diabetes and Atherosclerotic Cardiovascular Disease: Insights from the TECOS Trial. Diabetes Care 2017, 40, 1763–1770. [Google Scholar] [CrossRef] [PubMed]
- Barroso, M.; Goday, A.; Ramos, R.; Marin-Ibanez, A.; Guembe, M.J.; Rigo, F.; Tormo-Diaz, M.J.; Moreno-Iribas, C.; Cabre, J.J.; Segura, A.; et al. Interaction between cardiovascular risk factors and body mass index and 10-year incidence of cardiovascular disease, cancer death, and overall mortality. Prev. Med. 2018, 107, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Pagidipati, N.J.; Gaziano, T.A. Estimating Deaths from Cardiovascular Disease: A Review of Global Methodologies of Mortality Measurement. Circulation 2013, 127, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Nikiforov, N.G.; Elizova, N.V.; Orekhov, A.N. Macrophages and Their Contribution to the Development of Atherosclerosis. Results Probl. Cell Differ. 2017, 62, 273–298. [Google Scholar] [PubMed]
- Cochain, C.; Zernecke, A. Macrophages in vascular inflammation and atherosclerosis. Pflugers Arch. 2017, 469, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Bugge, A.; El-Naaman, B.; McMurray, R.G.; Froberg, K.; Nielsen, C.H.; Muller, K.; Andersen, L.B. Inflammatory markers and clustered cardiovascular disease risk factors in Danish adolescents. Horm. Res. Paediatr. 2012, 78, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Adib-Conquy, M.; Monchi, M.; Goulenok, C.; Laurent, I.; Thuong, M.; Cavaillon, J.M.; Adrie, C. Increased plasma levels of soluble triggering receptor expressed on myeloid cells 1 and procalcitonin after cardiac surgery and cardiac arrest without infection. Shock 2007, 28, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Buckland, K.F.; Ramaprakash, H.; Murray, L.A.; Carpenter, K.J.; Choi, E.S.; Kunkel, S.L.; Lukacs, N.W.; Xing, Z.; Aoki, N.; Hartl, D.; et al. Triggering receptor expressed on myeloid cells-1 (TREM-1) modulates immune responses to aspergillus fumigatus during fungal asthma in mice. Immunol. Investig. 2011, 40, 692–722. [Google Scholar] [CrossRef] [PubMed]
- Campanholle, G.; Mittelsteadt, K.; Nakagawa, S.; Kobayashi, A.; Lin, S.L.; Gharib, S.A.; Heinecke, J.W.; Hamerman, J.A.; Altemeier, W.A.; Duffield, J.S. TLR-2/TLR-4 TREM-1 signaling pathway is dispensable in inflammatory myeloid cells during sterile kidney injury. PLoS ONE 2013, 8, e68640. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.H.; Berim, I.G.; Agrawal, D.K. Chronic inflammation and cancer: Emerging roles of triggering receptors expressed on myeloid cells. Expert Rev. Clin. Immunol. 2015, 11, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Hyder, L.A.; Gonzalez, J.; Harden, J.L.; Johnson-Huang, L.M.; Zaba, L.C.; Pierson, K.C.; Eungdamrong, N.J.; Lentini, T.; Gulati, N.; Fuentes-Duculan, J.; et al. TREM-1 as a Potential Therapeutic Target in Psoriasis. J. Investig. Dermatol. 2013, 133, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Joffre, J.; Potteaux, S.; Zeboudj, L.; Loyer, X.; Boufenzer, A.; Laurans, L.; Esposito, B.; Vandestienne, M.; de Jager, S.C.; Hénique, C.; et al. Genetic and Pharmacological Inhibition of TREM-1 Limits the Development of Experimental Atherosclerosis. J. Am. Coll. Cardiol. 2016, 68, 2776–2793. [Google Scholar] [CrossRef] [PubMed]
- Lemarié, J.; Barraud, D.; Gibot, S. Host response biomarkers in sepsis: Overview on sTREM-1 detection. Methods Mol. Biol. 2015, 1237, 225–239. [Google Scholar] [PubMed]
- Lemarie, J.; Boufenzer, A.; Popovic, B.; Tran, N.; Groubatch, F.; Derive, M.; Labroca, P.; Barraud, D.; Gibot, S. Pharmacological inhibition of the triggering receptor expressed on myeloid cells-1 limits reperfusion injury in a porcine model of myocardial infarction. ESC Heart Fail. 2015, 2, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boufenzer, A.; Lemarie, J.; Simon, T.; Derive, M.; Bouazza, Y.; Tran, N.; Maskali, F.; Groubatch, F.; Bonnin, P.; Bastien, C.; et al. TREM-1 Mediates Inflammatory Injury and Cardiac Remodeling Following Myocardial Infarction. Circul. Res. 2015, 116, 1772–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Facchetti, F. TREM-1 (triggering receptor expressed on myeloid cells): A new player in acute inflammatory responses. J. Infect. Dis. 2003, 187 (Suppl. 2), S397–S401. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.; Joosten, L.A.; Dinarello, C.A.; Kullberg, B.J.; van der Meer, J.W.; Netea, M.G. TREM-1 interaction with the LPS/TLR4 receptor complex. Eur. Cytokine Netw. 2011, 22, 11–14. [Google Scholar] [PubMed]
- Ichou, L.; Carbonell, N.; Rautou, P.E.; Laurans, L.; Bourcier, S.; Pichereau, C.; Baudel, J.L.; Nousbaum, J.B.; Renou, C.; Anty, R.; et al. Ascitic fluid TREM-1 for the diagnosis of spontaneous bacterial peritonitis. Gut 2016, 65, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Lai, C.; Fife, M.E.; Grabiec, A.M.; Tildy, B.; Snelgrove, R.J.; Xin, G.; Lloyd, C.M.; Hussell, T. Reversal of TREM-1 ectodomain shedding and improved bacterial clearance by intranasal metalloproteinase inhibitors. Mucosal Immunol. 2017, 10, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Billioud, V.; Gibot, S.; Massin, F.; Oussalah, A.; Chevaux, J.B.; Williet, N.; Bronowicki, J.P.; Bigard, M.A.; Guéant, J.L.; Peyrin-Biroulet, L. Plasma soluble triggering receptor expressed on myeloid cells-1 in Crohn’s disease. Dig. Liver Dis. 2012, 44, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Gibot, S.; Massin, F.; Le Renard, P.; Béné, M.C.; Faure, G.C.; Bollaert, P.E.; Levy, B. Surface and soluble triggering receptor expressed on myeloid cells-1: Expression patterns in murine sepsis. Crit. Care Med. 2005, 33, 1787–1793. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Piña, V.; Soares-Schanoski, A.; Rodríguez-Rojas, A.; Fresno, C.D.; García, F.; Vallejo-Cremades, M.T.; Fernández-Ruiz, I.; Arnalich, F.; Fuentes-Prior, P.; López-Collazo, E. Metalloproteinases Shed TREM-1 Ectodomain from Lipopolysaccharide-Stimulated Human Monocytes. J. Immunol. 2007, 179, 4065–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchon, A.; Hernández-Munain, C.; Cella, M.; Colonna, M. A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 2001, 194, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting edge: Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef] [PubMed]
- Bouchon, A.; Facchetti, F.; Weigand, M.A.; Colonna, M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 2001, 410, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Gibot, S.; Alauzet, C.; Massin, F.; Sennoune, N.; Faure, G.C.; Béné, M.C.; Lozniewski, A.; Bollaert, P.E.; Lévy, B. Modulation of the triggering receptor expressed on myeloid cells-1 pathway during pneumonia in rats. J. Infect. Dis. 2006, 194, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. TREM-1: A new regulator of innate immunity in sepsis syndrome. Nat. Med. 2001, 7, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G. TREM-1: Intracellular signaling pathways and interaction with pattern recognition receptors. J. Leukocyte Biol. 2013, 93, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Dower, K.; Ellis, D.; Saraf, K.; Jelinsky, S.A.; Lin, L.-L. Innate Immune Responses to TREM-1 Activation: Overlap, Divergence, and Positive and Negative Cross-Talk with Bacterial Lipopolysaccharide. J. Immunol. 2008, 180, 3520–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Azam, T.; Ferwerda, G.; Girardin, S.E.; Kim, S.H.; Dinarello, C.A. Triggering receptor expressed on myeloid cells-1 (TREM-1) amplifies the signals induced by the NACHT-LRR (NLR) pattern recognition receptors. J. Leukocyte Biol. 2006, 80, 1454–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, S.C.; Agrawal, D.K. Toll-like receptors, triggering receptor expressed on myeloid cells family members and receptor for advanced glycation end-products in allergic airway inflammation. Expert Rev. Respir. Med. 2016, 10, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Pallati, P.K.; Rai, V.; Sharma, P.; Agrawal, D.K.; Nandipati, K.C. Increased expression of triggering receptor expressed on myeloid cells-1 in the population with obesity and insulin resistance. Obesity 2017, 25, 527–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thankam, F.G.; Dilisio, M.F.; Dougherty, K.A.; Dietz, N.E.; Agrawal, D.K. Triggering receptor expressed on myeloid cells and 5’adenosine monophosphate-activated protein kinase in the inflammatory response: A potential therapeutic target. Expert Rev. Clin. Immunol. 2016, 12, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, J.; Salcedo, R.; Mivechi, N.F.; Trinchieri, G.; Horuzsko, A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012, 72, 3977–3986. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Schiraldi, M.; Uguccioni, M.; Bianchi, M.E. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 2013, 55, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Casalgrandi, M.; Schiraldi, M.; Antoine, D.J.; Cattaneo, A.; De Marchis, F.; Liu, J.; Antonelli, A.; Preti, A.; Raeli, L.; et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 2012, 209, 1519–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiraldi, M.; Raucci, A.; Munoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; De Marchis, F.; Pedotti, M.; Bachi, A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 2012, 209, 551–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bingold, T.M.; Pullmann, B.; Sartorius, S.; Geiger, E.V.; Marzi, I.; Zacharowski, K.; Wissing, H.; Scheller, B. Soluble triggering receptor on myeloid cells-1 is expressed in the course of non-infectious inflammation after traumatic lung contusion: A prospective cohort study. Crit. Care 2011, 15, R115. [Google Scholar] [CrossRef] [PubMed]
- Brynjolfsson, S.F.; Magnusson, M.K.; Kong, P.L.; Jensen, T.; Kuijper, J.L.; Håkansson, K.; Read, C.B.; Stennicke, V.W.; Sjövall, H.; Jo Wick, M. An Antibody against Triggering Receptor Expressed on Myeloid Cells 1 (TREM-1) Dampens Proinflammatory Cytokine Secretion by Lamina Propria Cells from Patients with IBD. Inflamm. Bowel Dis. 2016, 22, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Radsak, M.P.; Taube, C.; Haselmayer, P.; Tenzer, S.; Salih, H.R.; Wiewrodt, R.; Buhl, R.; Schild, H. Soluble triggering receptor expressed on myeloid cells 1 is released in patients with stable chronic obstructive pulmonary disease. Clin. Dev. Immunol. 2007, 2007, 52040. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Yang, Z.-Y.; Zhang, X.; Wu, H.-J. Molecular cloning and mRNA expression of the peptidoglycan recognition protein gene HcPGRP1 and its isoform HcPGRP1a from the freshwater mussel Hyriopsis cumingi. Genet. Mol. Biol. 2014, 37, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, A.; Neyen, C.; Lemaitre, B.; Marion-Poll, F.; Skoulakis, E.M.C. The gram-negative sensing receptor PGRP-LC contributes to grooming induction in Drosophila. PLoS ONE 2017, 12, e0185370. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.-Y.; Yang, P.-J.; Rao, X.-J. Molecular cloning and analysis of PGRP-L1 and IMD from silkworm Bombyx mori. Comp. Biochem. Physiol. B 2018, 215, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Ornatowska, M.; Joo, M.S.; Sadikot, R.T. TREM-1 expression in macrophages is regulated at transcriptional level by NF-κB and PU.1. Eur. J. Immunol. 2007, 37, 2300–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tintinger, G.R.; van der Merwe, J.J.; Fickl, H.; Rheeder, P.; Feldman, C.; Anderson, R. Soluble triggering receptor expressed on myeloid cells in sputum of patients with community-acquired pneumonia or pulmonary tuberculosis: A pilot study. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Luyt, C.E.; Combes, A.; Trouillet, J.L.; Chastre, J. Biomarkers to optimize antibiotic therapy for pneumonia due to multidrug-resistant pathogens. Clin. Chest Med. 2011, 32, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Liu, Z.; Wei, S.; Xu, F.; Chen, Z.; Gong, J. Triggering receptor in myeloid cells (TREM-1) specific expression in peripheral blood mononuclear cells of sepsis patients with acute cholangitis. Inflammation 2009, 32, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, A.; Kers, J.; Emal, D.; Stroo, I.; Teske, G.J.; Butter, L.M.; Claessen, N.; Damman, J.; Derive, M.; Navis, G.J.; et al. Corrigendum: Effect of TREM-1 blockade and single nucleotide variants in experimental renal injury and kidney transplantation. Sci. Rep. 2017, 7, 44163. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Pallati, P.K.; Sharma, P.; Agrawal, D.K.; Nandipati, K.C. Significant association of TREM-1 with HMGB1, TLRs and RAGE in the pathogenesis of insulin resistance in obese diabetic populations. Am. J. Transl. Res. 2017, 9, 3224–3244. [Google Scholar] [PubMed]
- Subramanian, S.; Pallati, P.K.; Sharma, P.; Agrawal, D.K.; Nandipati, K.C. TREM-1 associated macrophage polarization plays a significant role in inducing insulin resistance in obese population. J. Transl. Med. 2017, 15, 85. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, A.; Stroo, I.; Rampanelli, E.; Blank, F.; Butter, L.M.; Claessen, N.; Takai, T.; Colonna, M.; Leemans, J.C.; Florquin, S.; et al. Role of TREM1-DAP12 in renal inflammation during obstructive nephropathy. PLoS ONE 2013, 8, e82498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golovkin, A.S.; Ponasenko, A.V.; Yuzhalin, A.E.; Salakhov, R.R.; Khutornaya, M.V.; Kutikhin, A.G.; Rutkovskaya, N.V.; Savostyanova, Y.Y.; Barbarash, L.S. An association between single nucleotide polymorphisms within TLR and TREM-1 genes and infective endocarditis. Cytokine 2015, 71, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.; Bouchon, A.; Seibold, F.; Mueller, C. TREM-1—Expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J. Clin. Investig. 2007, 117, 3097–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thankam, F.G.; Dilisio, M.F.; Dietz, N.E.; Agrawal, D.K. TREM-1, HMGB1 and RAGE in the Shoulder Tendon: Dual Mechanisms for Inflammation Based on the Coincidence of Glenohumeral Arthritis. PLoS ONE 2016, 11, e0165492. [Google Scholar] [CrossRef] [PubMed]
- Joffre, J.; Ait-Oufella, H. Targeting the immune response in atherosclerosis: It’s time for clinical trials! Arch. Cardiovasc. Dis. 2017, 110, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Birkenheuer, A.J.; Marr, H.S.; Levy, M.G.; Yoder, J.A.; Nordone, S.K. Expression and function of triggering receptor expressed on myeloid cells-1 (TREM-1) on canine neutrophils. Dev. Comp. Immunol. 2011, 35, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Rao, V.H.; Shao, Z.; Agrawal, D.K. Dendritic Cells Expressing Triggering Receptor Expressed on Myeloid Cells-1 Correlate with Plaque Stability in Symptomatic and Asymptomatic Patients with Carotid Stenosis. PLoS ONE 2016, 11, e0154802. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.H.; Rai, V.; Stoupa, S.; Subramanian, S.; Agrawal, D.K. Data on TREM-1 activation destabilizing carotid plaques. Data Brief 2016, 8, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Golovkin, A.S.; Ponasenko, A.V.; Khutornaya, M.V.; Kutikhin, A.G.; Salakhov, R.R.; Yuzhalin, A.E.; Zhidkova, I.I.; Barbarash, O.L.; Barbarash, L.S. Association of TLR and TREM-1 gene polymorphisms with risk of coronary artery disease in a Russian population. Gene 2014, 550, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Ponasenko, A.V.; Khutornaya, M.V.; Yuzhalin, A.E.; Zhidkova, I.I.; Salakhov, R.R.; Golovkin, A.S.; Barbarash, O.L.; Barbarash, L.S. Association of TLR and TREM-1 gene polymorphisms with atherosclerosis severity in a Russian population. Meta Gene 2016, 9, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, C.; Ding, F.H.; Shen, Y.; Gao, J.; Liu, Z.H.; Chen, J.W.; Zhang, R.Y.; Shen, W.F.; Wang, X.Q.; et al. Data on the expression and role of TREM-1 in the development of in-stent restenosis. Data Brief 2018, 16, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, C.; Ding, F.H.; Shen, Y.; Gao, J.; Liu, Z.H.; Chen, J.W.; Zhang, R.Y.; Shen, W.F.; Wang, X.Q.; et al. Increased serum TREM-1 level is associated with in-stent restenosis, and activation of TREM-1 promotes inflammation, proliferation and migration in vascular smooth muscle cells. Atherosclerosis 2017, 267, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Schiechl, G.; Brunner, S.M.; Kesselring, R.; Martin, M.; Ruemmele, P.; Mack, M.; Hirt, S.W.; Schlitt, H.J.; Geissler, E.K.; Fichtner-Feigl, S. Inhibition of Innate Co-Receptor TREM-1 Signaling Reduces CD4(+) T Cell Activation and Prolongs Cardiac Allograft Survival. Am. J. Transplant. 2013, 13, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hong, F.; Pan, S.; Lei, L.; Yan, F. Silencing Triggering Receptors Expressed on Myeloid Cells-1 Impaired the Inflammatory Response to Oxidized Low-Density Lipoprotein in Macrophages. Inflammation 2016, 39, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.H.; Rai, V.; Stoupa, S.; Subramanian, S.; Agrawal, D.K. Tumor necrosis factor-alpha regulates triggering receptor expressed on myeloid cells-1-dependent matrix metalloproteinases in the carotid plaques of symptomatic patients with carotid stenosis. Atherosclerosis 2016, 248, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Arguinano, A.A.A.; Dade, S.; Stathopoulou, M.; Derive, M.; Ndiaye, N.C.; Xie, T.; Masson, C.; Gibot, S.; Visvikis-Siest, S. TREM-1 SNP rs2234246 regulates TREM-1 protein and mRNA levels and is associated with plasma levels of l-selectin. PLoS ONE 2017, 12, e0182226. [Google Scholar]
- Jeremie, L.; Amir, B.; Marc, D.; Sebastien, G. The Triggering Receptor Expressed on Myeloid cells-1: A new player during acute myocardial infarction. Pharmacol. Res. 2015, 100, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Lemarie, J.; Boufenzer, A.; Derive, M.; Maskali, F.; Marie, P.-Y.; Gibot, S. Protective effects of TREM-1 modulation during experimental myocardial infarction. Eur. Heart J. 2013, 34, 3691. [Google Scholar] [CrossRef]
- Tao, F.; Peng, L.; Li, J.; Shao, Y.; Deng, L.; Yao, H. Association of serum myeloid cells of soluble triggering receptor-1 level with myocardial dysfunction in patients with severe sepsis. Mediat. Inflamm. 2013, 2013, 819246. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Ye, L.; Zhang, L.; Zhang, Y.; Deng, L.; Yao, H. Association of myeloid cells of triggering receptor-1 with left ventricular systolic dysfunction in BALB/c mice with sepsis. Mediat. Inflamm. 2014, 2014, 391492. [Google Scholar] [CrossRef] [PubMed]
- Ponasenko, A.V.; Kutikhin, A.G.; Khutornaya, M.V.; Yuzhalin, A.E.; Rutkovskaya, N.V.; Golovkin, A.S. Association of Trem-1 Gene Polymorphisms with Infective Endocarditis. Infektsiya I Immunitet 2015, 5, 331–338. [Google Scholar] [CrossRef]
- Rosenfeld, M.E.; Campbell, L.A. Pathogens and atherosclerosis: Update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [PubMed]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Polo, M.T.; Castells, M.T.; García-Pérez, B.; Martín, A.; Adánez, G.; Ayala, I. Effect of diet/atorvastatin on atherosclerotic lesions associated to nonalcoholic fatty liver disease in chickens. Histol. Histopathol. 2015, 30, 1439–1446. [Google Scholar] [PubMed]
- Rai, V.; Agrawal, D.K. The role of damage- and pathogen-associated molecular patterns in inflammation-mediated vulnerability of atherosclerotic plaques. Can. J. Physiol. Pharmacol. 2017, 95, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankenberg, S.; Barbaux, S.; Tiret, L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003, 170, 191–203. [Google Scholar] [CrossRef]
- Wildgruber, M.; Czubba, M.; Aschenbrenner, T.; Wendorff, H.; Hapfelmeier, A.; Glinzer, A.; Schiemann, M.; Zimmermann, A.; Eckstein, H.H.; Berger, H.; et al. Increased intermediate CD14(++)CD16(++) monocyte subset levels associate with restenosis after peripheral percutaneous transluminal angioplasty. Atherosclerosis 2016, 253, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Vlodaver, Z.; Wilson, R.F. Complications of Acute Myocardial Infarction; Springer: New York, NY, USA, 2012; pp. 321–347. [Google Scholar]
- Akbar, N.; Digby, J.E.; Cahill, T.J.; Tavare, A.N.; Saluja, S.; Dawkins, S.; Edgar, L.; Rawlings, N.; Ziberna, K.; McNeill, E.; et al. Endothelium-derived extracellular vesicles promote splenic monocyte mobilization in myocardial infarction. JCI Insight 2017, 2, 93344. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, N.; Godec, J.; Lee, R.; Chai, J.T.; Dall’Armellina, E.; McAndrew, D.J.; Digby, J.E.; Forfar, J.C.; Prendergast, B.; Kharbanda, R.K.; et al. Acute myocardial infarction activates distinct inflammation and proliferation pathways in circulating monocytes, prior to recruitment, and identified through conserved transcriptional responses in mice and humans. Eur. Heart J. 2015, 36, 1923–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Oh, S.S.; Lim, D.S.; Na, C.Y.; Kim, J.H. Clinical significance of cerebrovascular complications in patients with acute infective endocarditis: A retrospective analysis of a 12-year single-center experience. BMC Neurol. 2014, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Zhang, H.; Qu, W.; Lv, J.; Wang, Y.; Xiao, J. Increased sTREM-1 in pregnant women with premature rupture of membranes and subclinical chorioamnionitis. Mol. Med. Rep. 2012, 5, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, P.; Jashari, F.; Nicoll, R.; Bajraktari, G.; Wester, P.; Henein, M.Y. Coronary and carotid atherosclerosis: How useful is the imaging? Atherosclerosis 2013, 231, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Gibot, S.; Kolopp-Sarda, M.N.; Béné, M.C.; Bollaert, P.E.; Lozniewski, A.; Mory, F.; Levy, B.; Faure, G.C. A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J. Exp. Med. 2004, 200, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Derive, M.; Bouazza, Y.; Sennoun, N.; Marchionni, S.; Quigley, L.; Washington, V.; Massin, F.; Max, J.P.; Ford, J.; Alauzet, C.; et al. Soluble TREM-like transcript-1 regulates leukocyte activation and controls microbial sepsis. J. Immunol. 2012, 188, 5585–5592. [Google Scholar] [CrossRef] [PubMed]
- Pelham, C.J.; Agrawal, D.K. Emerging roles for triggering receptor expressed on myeloid cells receptor family signaling in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Derive, M.; Massin, F.; Gibot, S. Triggering receptor expressed on myeloid cells-1 as a new therapeutic target during inflammatory diseases. Self/Nonself 2010, 1, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Derive, M.; Boufenzer, A.; Gibot, S. Attenuation of Responses to Endotoxin by the Triggering Receptor Expressed on Myeloid Cells-1 Inhibitor LR12 in Nonhuman Primate. Anesthesiology 2014, 120, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, K.; Boufenzer, A.; Jolly, L.; Cordier, H.L.; Wang, G.; Heck, A.J.R.; Cerwenka, A.; Vinolo, E.; Nazabal, A.; Kriznik, A.; et al. TREM-1 multimerization is essential for its activation on monocytes and neutrophils. Cell. Mol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Syed, M.A.; Panchal, D.; Rogers, D.; Joo, M.; Sadikot, R.T. Curcumin mediated epigenetic modulation inhibits TREM-1 expression in response to lipopolysaccharide. Int. J. Biochem. Cell Biol. 2012, 44, 2032–2043. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Kohsaka, H.; Kitasato, H.; Akahoshi, T. Lipopolysaccharide-Induced Up-Regulation of Triggering Receptor Expressed on Myeloid Cells-1 Expression on Macrophages Is Regulated by Endogenous Prostaglandin E2. J. Immunol. 2007, 178, 1144–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.T.; Sigalov, A.B. Novel TREM-1 Inhibitors Attenuate Tumor Growth and Prolong Survival in Experimental Pancreatic Cancer. Mol. Pharm. 2017, 14, 4572–4582. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B. A novel ligand-independent peptide inhibitor of TREM-1 suppresses tumor growth in human lung cancer xenografts and prolongs survival of mice with lipopolysaccharide-induced septic shock. Int. Immunopharmacol. 2014, 21, 208–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Guo, Y.; Jiang, Y.; Zhu, X.; Zhang, X. Vitamin D suppresses macrophage infiltration by down-regulation of TREM-1 in diabetic nephropathy rats. Mol. Cell. Endocrinol. 2018, 473, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zysset, D.; Weber, B.; Rihs, S.; Brasseit, J.; Freigang, S.; Riether, C.; Banz, Y.; Cerwenka, A.; Simillion, C.; Marques-Vidal, P.; et al. TREM-1 links dyslipidemia to inflammation and lipid deposition in atherosclerosis. Nat. Commun. 2016, 7, 13151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, K.; You, Y.; Swier, V.; Tang, L.; Radwan, M.M.; Pandya, A.N.; Agrawal, D.K. Vitamin D Protects Against Atherosclerosis via Regulation of Cholesterol Efflux and Macrophage Polarization in Hypercholesterolemic Swine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2432–2442. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CVD | Approach | Outcome | Model Ref |
|---|---|---|---|
| Atherosclerosis | Effect of oxLDL on TREM-1 expression in macrophages during atherogenesis | oxLDL increased TREM-1 expression and its interaction with TLR-4 to amplify inflammation. This action is reduced by TREM-1 silencing or inhibition | M [66] |
| Atherosclerosis | Role played by TREM-1 in macrophages’ involvement in atherosclerosis | TREM-1, in association with TLR-4, contributes to formation of foam cell derived from macrophages through inflammatory response | M [12] |
| Atherosclerosis | TERM-1 polymorphism is associated with atherosclerosis severity | rs4711668 polymorphism within TREM-1 gene and TLR-2 are associated coronary atherosclerosis, TLR-1,4,6 with mild coronary atherosclerosis. | H [61] |
| Atherosclerosis | TREM-1 expression on dendritic cells (DC) in atherosclerotic plaques | TREM-1 was upregulated in DCs of atherosclerotic plaque and was positively correlated with plaque destabilization | H [59] |
| Atherosclerosis | TREM-1 expression in vulnerability of atheroma plaque | Increased expression of TREM-1 in VSMCs is associated with plaque vulnerability | H [60,67] |
| Acute myocardial infarction | sTREM-1 level regulation by its polymorphism and plasma sl-selectins level | SNP (rs2234246) is associated with increased plasma sTREM-1 and l-selectin | H [68] |
| Acute myocardial infarction | Role played by TREM-1 in inflammatory response after AMI | TREM-1 deletion or inhibition decreases inflammation after AMI | M [69] |
| Acute myocardial infarction | TREM-1 inhibition by LR12’s effect on reperfusion injury after MI | TREM-1 inhibition by LR-12 amends the reperfusion injury of the myocardia | S [70] |
| Acute myocardial infarction | TREM-1 expression in causing innate immune and inflammatory responses after myocardial infarcts | TREM-1 genetic inhibition reduced inflammation and sTREM-1 level. TREM-1 is positively correlated with AMI severity | M [15] |
| Coronary artery diseases | TREM-1 and TLR polymorphisms in CAD | Polymorphisms in TREM-1 and in TLRs were robustly associated to CAD | H [61] |
| In-stent restenosis | Association of sTREM-1 with in-stent restenosis and expression of TREM-1 in VSMCs | sTREM-1 was elevated in patient with stent restenosis, and TREM-1 induces VSMCs inflammation, migration, and proliferation | H [64] |
| Myocardial dysfunction in septicemia | Association of level of sTREM-1 and severity of myocardial dysfunction in septicemia | sTREM level predicts myocardial dysfunction in septicemia | H [71] |
| Myocardial dysfunction | Association of TREM-1 with LPS-induced ventricular dysfunction | TREM-1 plays a significant role in LPS-induced ventricular dysfunction | M [72] |
| Infective Endocarditis | How polymorphism in TREM-1 and TLRs affects the outcome of IE | No association was found between SNPs within TREM-1 genes and the outcome of IE | H [54] |
| Infective Endocarditis | How heredity of TREM-1 variation could affect the susceptibility and outcome of IE | Only rs1817537 polymorphism is associated with high susceptibility to IE | H [73] |
| Cardiac transplant | How TREM-1 and antigen-presenting cells affect alloreactive CD4 and lymphocytes | TREM-1 contribute to the differentiation and proliferation of CD-4 positive lymphocytes | M [65] |
| Cardiac arrest after heart Surgery | sTREM-1 level after cardiac events without infection | TREM-1 along with procalcitonin increase during cardiac events and are not specific to infection but to inflammation | H [7] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouassi, K.T.; Gunasekar, P.; Agrawal, D.K.; Jadhav, G.P. TREM-1; Is It a Pivotal Target for Cardiovascular Diseases? J. Cardiovasc. Dev. Dis. 2018, 5, 45. https://doi.org/10.3390/jcdd5030045
Kouassi KT, Gunasekar P, Agrawal DK, Jadhav GP. TREM-1; Is It a Pivotal Target for Cardiovascular Diseases? Journal of Cardiovascular Development and Disease. 2018; 5(3):45. https://doi.org/10.3390/jcdd5030045
Chicago/Turabian StyleKouassi, Kouassi T., Palanikumar Gunasekar, Devendra K. Agrawal, and Gopal P. Jadhav. 2018. "TREM-1; Is It a Pivotal Target for Cardiovascular Diseases?" Journal of Cardiovascular Development and Disease 5, no. 3: 45. https://doi.org/10.3390/jcdd5030045
APA StyleKouassi, K. T., Gunasekar, P., Agrawal, D. K., & Jadhav, G. P. (2018). TREM-1; Is It a Pivotal Target for Cardiovascular Diseases? Journal of Cardiovascular Development and Disease, 5(3), 45. https://doi.org/10.3390/jcdd5030045