Hemodynamics in Cardiac Development


Abstract
1. Introduction
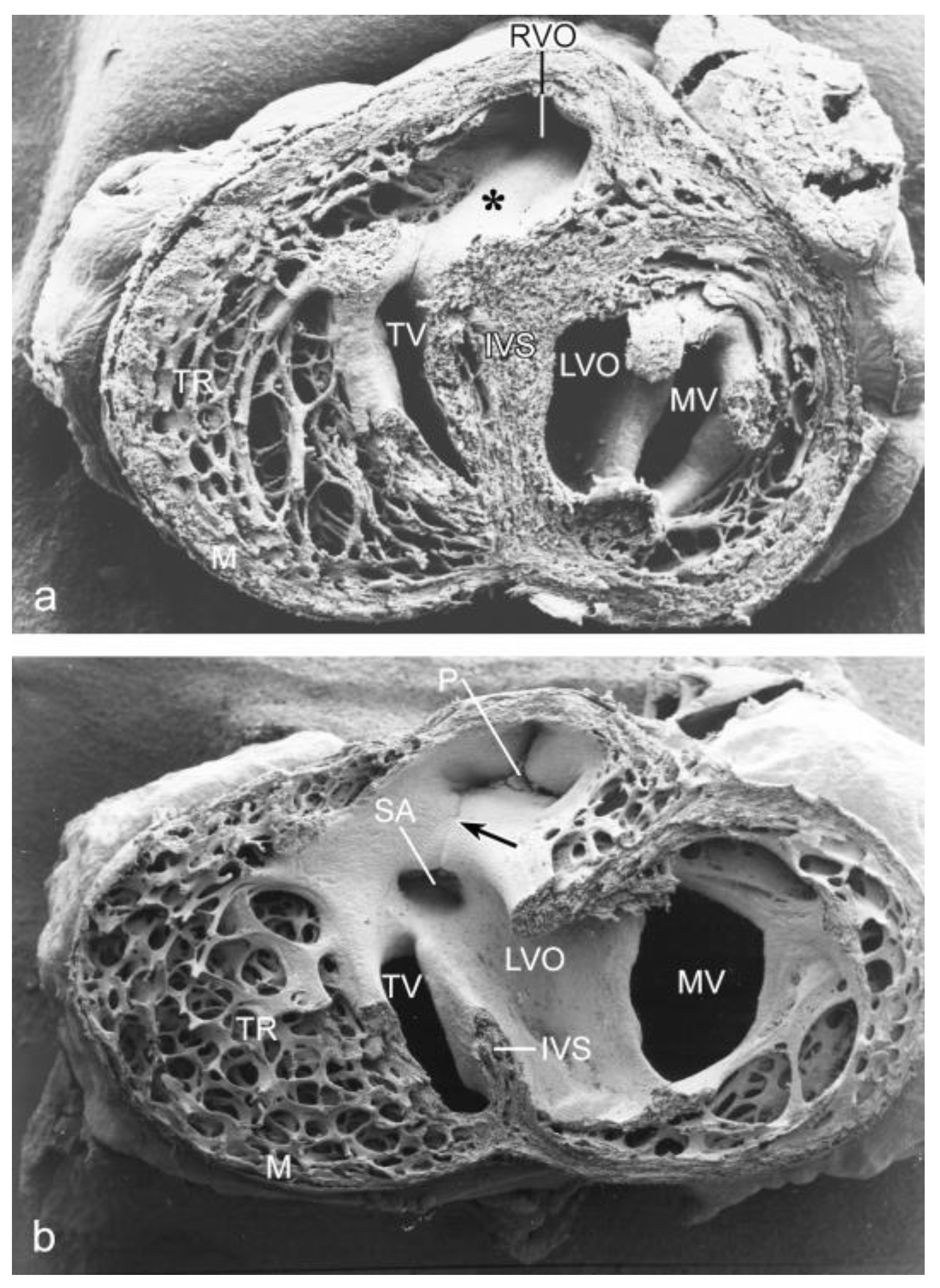
2. Cardiac Anomalies after Vitelline Vein Ligation
Materials and Methods
3. Results
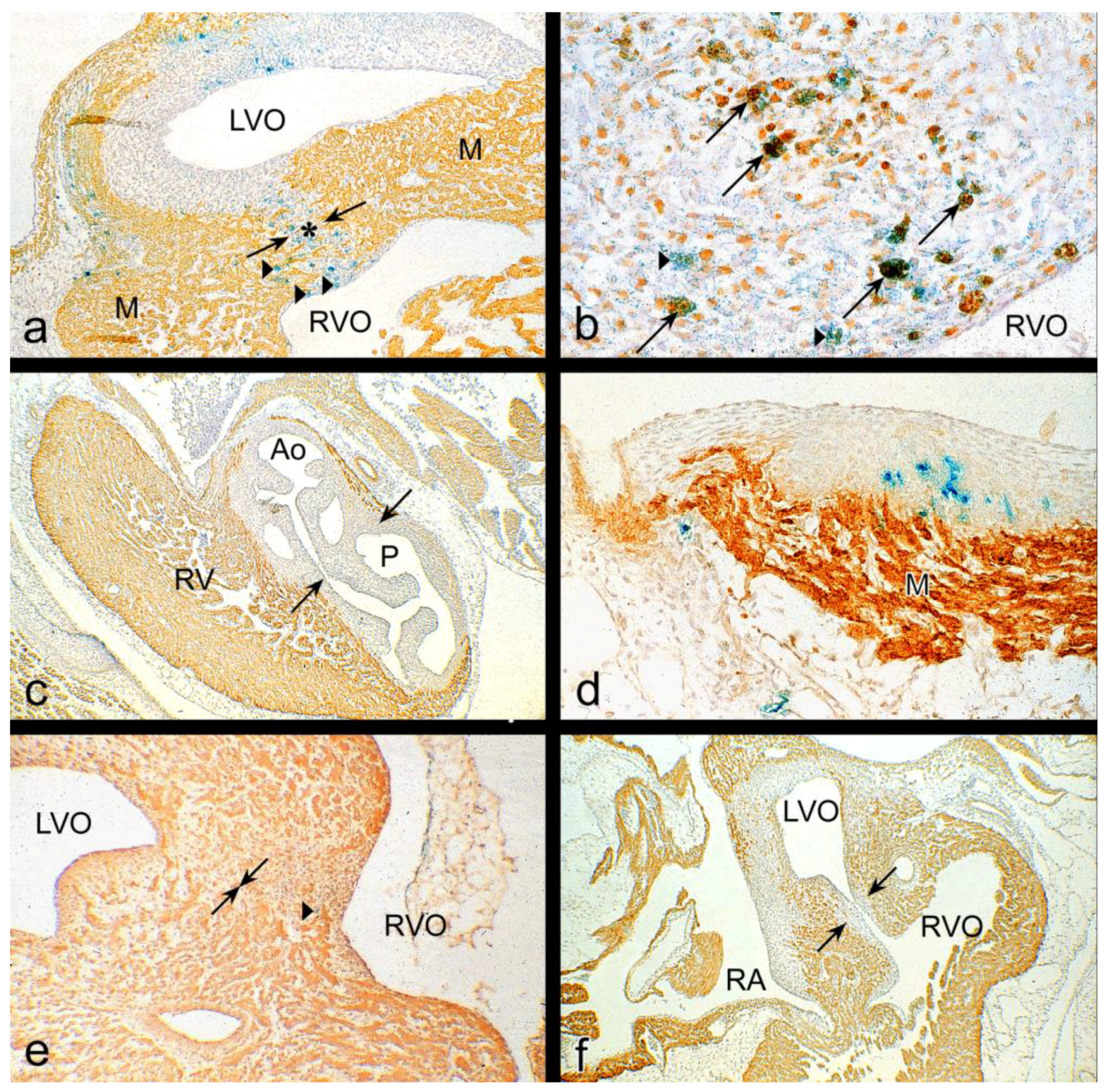
3.1. Impaired Development in Preseptation Stages and Tgfβ Receptor III (TBRIII) Expression
3.2. Peri- and Post-Septation Stages
4. Discussion
4.1. Hemodynamic Load
4.2. Ciliary Mechanosensing
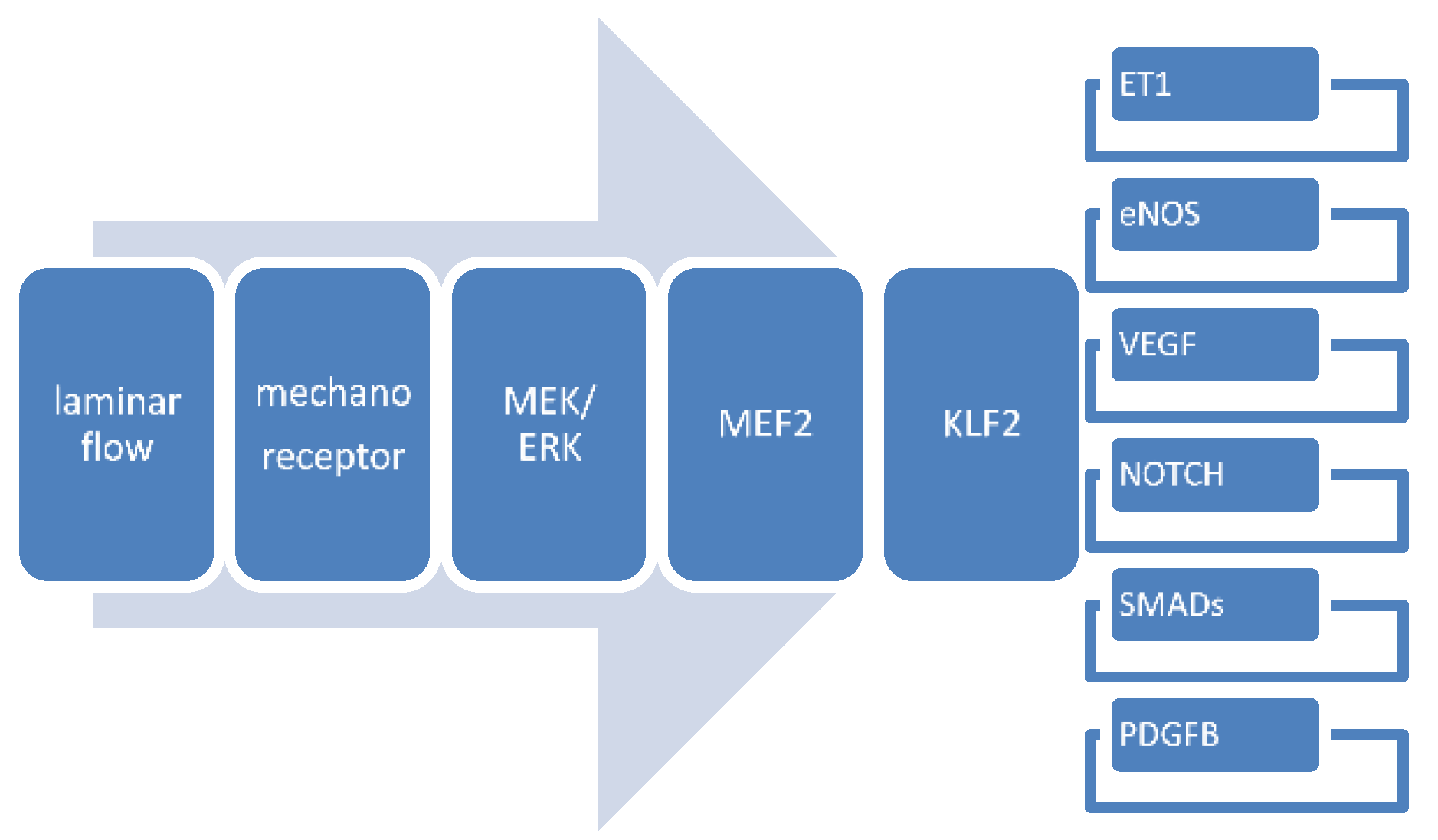
4.3. Gene Expression Patterns in Mechanosensing
4.3.1. Vascular Endothelial Growth Factor (Vegf) Signaling
4.3.2. Notch Signaling
4.3.3. Platelet-Derived Growth Factor (Pdgf) Signaling
4.3.4. Krüppel-Like Factor-2
4.3.5. Endothelin Signaling
4.3.6. Nitric Oxide (NO) Signaling
4.3.7. Tgfβ/Bmp Signaling
4.3.8. Interactions between Flow-Responsive Genes
4.4. Consequences for Human Embryonic Development
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gittenberger-de Groot, A.C.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R. Morphogenesis and molecular considerations on congenital cardiac septal defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Hogers, B.; DeRuiter, M.C.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Unilateral vitelline vein ligation alters intracardiac blood flow patterns and morphogenesis in the chick embryo. Circ. Res. 1997, 80, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, B.C.; Hierck, B.P.; Vrolijk, J.; Baiker, M.; Pourquie, M.J.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Changes in shear stress-related gene expression after experimentally altered venous return in the chicken embryo. Circ. Res. 2005, 96, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G. The second heart field. Curr. Top. Dev. Biol. 2012, 100, 33–65. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, B.C.; Van der Heiden, K.; Hierck, B.P.; Poelmann, R.E. The role of shear stress on ET-1, KLF2, and NOS-3 expression in the developing cardiovascular system of chicken embryos in a venous ligation model. Physiology 2007, 22, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Okagawa, H.; Markwald, R.R.; Sugi, Y. Functional BMP receptor in endocardial cells is required in atrioventricular cushion mesenchymal cell formation in chick. Dev. Biol. 2007, 306, 179–928. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.P.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pomares, J.M.; Phelps, A.; Sedmerova, M.; Carmona, R.; González-Iriarte, M.; Muñoz-Chápuli, R.; Wessels, A. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: A model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 2002, 247, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Poelmann, R.E.; Mikawa, T.; Gittenberger-de Groot, A.C. Neural crest cells in outflow tract septation of the embryonic chicken heart: Differentiation and apoptosis. Dev. Dyn. 1998, 212, 373–384. [Google Scholar] [CrossRef]
- Waldo, K.; Miyagawa-Tomita, S.; Kumiski, D.; Kirby, M.L. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: Aortic sac to ventricular septal closure. Dev. Biol. 1998, 196, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Goddeeris, M.M.; Schwartz, R.; Klingensmith, J.; Meyers, E.N. Independent requirements for Hedgehog signaling by both the anterior heart field and neural crest cells for outflow tract development. Development 2007, 134, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.C.; Chughtai, M.; Wisse, L.J.; Gittenberger-de Groot, A.C.; Feng, Q.; Goumans, M.T.H.; VanMunsteren, J.C.; Jongbloed, M.R.M.; DeRuiter, M.C. Nos3 mutation leads to abnormal neural crest cell and second heart field lineage patterning in bicuspid aortic valve formation. Dis. Model. Mech. 2018, Dmm.034637. [Google Scholar] [CrossRef]
- Gong, H.; Lyu, X.; Wang, Q.; Hu, M.; Zhang, X. Endothelial to mesenchymal transition in the cardiovascular system. Life Sci. 2017, 184, 95–102. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Agbu, S.; Anderson, K.V. Microtubule Motors Drive Hedgehog Signaling in Primary Cilia. Trends Cell Biol. 2017, 27, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Hildreth, V.; Webb, S.; Chaudhry, B.; Peat, J.D.; Phillips, H.M.; Brown, N.; Anderson, R.H.; Henderson, D.J. Left cardiac isomerism in the Sonic hedgehog null mouse. J. Anat. 2009, 214, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Shiratori, H.; Hamada, H. Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 2007, 450, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Hogers, B.; DeRuiter, M.C.; Baasten, A.M.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Intracardiac blood flow patterns related to the yolk sac circulation of the chick embryo. Circ. Res. 1995, 76, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Rugonyi, S.; Shaut, C.; Liu, A.; Thornburg, K.; Wang, R. Changes in wall motion and blood flow in the outflow tract of chick embryonic hearts observed with optical coherence tomography after outflow tract banding and vitelline-vein ligation. Phys. Med. Biol. 2008, 53, 5077–5091. [Google Scholar] [CrossRef] [PubMed]
- Poelma, C.; Van der Heiden, K.; Hierck, B.P.; Poelmann, R.E.; Westerweel, J. Measurements of the wall shear stress distribution in the outflow tract of an embryonic chicken heart. J. R. Soc. Interface 2010, 7, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, W.J.; Teslovich, N.C.; Menon, P.G.; Tinney, J.P.; Keller, B.B.; Pekkan, K. Left atrial ligation alters intracardiac flow patterns and the biomechanical landscape in the chick embryo. Dev. Dyn. 2014, 243, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Jamison, R.A.; Samarage, C.R.; Bryson-Richardson, R.J.; Fouras, A. In vivo wall shear measurements within the developing zebrafish heart. PLoS ONE 2013, 8, e75722. [Google Scholar] [CrossRef] [PubMed]
- Kalogirou, S.; Malissovas, N.; Moro, E.; Argenton, F.; Stainier, D.Y.; Beis, D. Intracardiac flow dynamics regulate atrioventricular valve morphogenesis. Cardiovasc. Res. 2014, 104, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Townsend, T.A.; Robinson, J.Y.; How, T.; DeLaughter, D.M.; Blobe, G.C.; Barnett, J.V. Endocardial cell epithelial-mesenchymal transformation requires Type III TGFβ receptor interaction with GIPC. Cell Signal. 2012, 24, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.D.; Van der Heiden, K.; Van de Pas, S.; Vennemann, P.; Poelma, C.; DeRuiter, M.C.; Goumans, M.J.; Gittenberger-de Groot, A.C.; Ten Dijke, P.; Poelmann, R.E.; et al. Tgfβ/Alk5 signaling is required for shear stress induced klf2 expression in embryonic endothelial cells. Dev. Dyn. 2011, 240, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Milgrom-Hoffman, M.; Michailovici, I.; Ferrara, N.; Zelzer, E.; Tzahor, E. Endothelial cells regulate neural crest and second heart field morphogenesis. Biol. Open 2014, 3, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Watanabe, Y.; Smyth, G.; Miyagawa-Tomita, S.; Meyers, E.; Klingensmith, J.; Camenisch, T.; Buckingham, M.; Moon, A.M. An FGF autocrine loop initiated in second heart field mesoderm regulates morphogenesis at the arterial pole of the heart. Development 2008, 135, 3599–3610. [Google Scholar] [CrossRef] [PubMed]
- Piek, E.; Heldin, C.H.; Ten Dijke, P. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999, 13, 2105–2124. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Bartram, U.; Molin, D.G.; Wisse, L.J.; Mohamad, A.; Sanford, L.P.; Doetschman, T.; Speer, C.P.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation 2001, 103, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.B.; Boyer, A.S.; Runyan, R.B.; Barnett, J.V. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science 1999, 283, 2080–2082. [Google Scholar] [CrossRef] [PubMed]
- Broekhuizen, M.L.; Hogers, B.; DeRuiter, M.C.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Wladimiroff, J.W. Altered hemodynamics in chick embryos after extraembryonic venous obstruction. Ultrasound Obstet. Gynecol. 1999, 13, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.H.; Chaudhry, B.; Mohun, T.J.; Bamforth, S.D.; Hoyland, D.; Phillips, H.M.; Webb, S.; Moorman, A.F.; Brown, N.A.; Henderson, D.J. Normal and abnormal development of the intrapericardial arterial trunks in humans and mice. Cardiovasc. Res. 2012, 95, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [PubMed]
- Bartelings, M.M.; Gittenberger-de Groot, A.C. The outflow tract of the heart-embryologic and morphologic correlations. Int. J. Cardiol. 1989, 22, 289–300. [Google Scholar] [CrossRef]
- Poelmann, R.E.; Molin, D.; Wisse, L.J.; Gittenberger-de Groot, A.C. Apoptosis in cardiac development. Cell Tissue Res. 2000, 301, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Brauer, P.R.; Yee, J.A. Cranial neural crest cells synthesize and secrete a latent form of transforming growth factor beta that can be activated by neural crest cell proteolysis. Dev. Biol. 1993, 155, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Brauer, P.R. Latent transforming growth factor-beta is present in the extracellular matrix of embryonic hearts in situ. Dev. Dyn. 1996, 205, 126–134. [Google Scholar] [CrossRef]
- Ramsdell, A.F.; Markwald, R.R. Induction of endocardial cushion tissue in the avian heart is regulated, in part, by TGFbeta-3-mediated autocrine signaling. Dev. Biol. 1997, 188, 64–74. [Google Scholar] [CrossRef] [PubMed]
- MacLellan, W.R.; Brand, T.; Schneider, M.D. Transforming growth factor-beta in cardiac ontogeny and adaptation. Circ. Res. 1993, 73, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Takamura, K.; Okishima, T.; Ohdo, S.; Hayakawa, K. Association of cephalic neural crest cells with cardiovascular development, particularly that of the semilunar valves. Anat. Embryol. 1990, 182, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, H.C.; Shekhar, A.; McQuinn, T.C.; Butcher, J.T. Hemodynamic patterning of the avian atrioventricular valve. Dev. Dyn. 2011, 240, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.E.; Butcher, J.T.; Yalcin, H.C. Mechanical regulation of cardiac development. Front. Physiol. 2014, 5, 318. [Google Scholar] [CrossRef] [PubMed]
- Midgett, M.; Rugonyi, S. Congenital heart malformations induced by hemodynamic altering surgical interventions. Front. Physiol. 2014, 5, 287. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, D.; Pexieder, T.; Rychterova, V.; Hu, N.; Clark, E.B. Remodeling of chick embryonic ventricular myoarchitecture under experimentally changed loading conditions. Anat. Rec. 1999, 254, 238–252. [Google Scholar] [CrossRef]
- Pesevski, Z.; Kvasilova, A.; Stopkova, T.; Nanka, O.; Drobna Krejci, E.; Buffinton, C.; Kockova, R.; Eckhardt, A.; Sedmera, D. Endocardial Fibroelastosis is Secondary to Hemodynamic Alterations in the Chick Embryonic Model of Hypoplastic Left Heart Syndrome. Dev. Dyn. 2018, 247, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Christensen, D.A.; Agrawal, A.K.; Beaumont, C.; Clark, E.B.; Hawkins, J.A. Dependence of aortic arch morphogenesis on intracardiac blood flow in the left atrial ligated chick embryo. Anat. Rec. 2009, 292, 652–660. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, A.; McQuinn, T.; Sedmera, D. Increased ventricular preload is compensated by myocyte proliferation in normal and hypoplastic fetal chick left ventricle. Circ. Res. 2007, 100, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Tobita, K.; Garrison, J.B.; Liu, L.J.; Tinney, J.P.; Keller, B.B. Three-dimensional myofiber architecture of the embryonic left ventricle during normal development and altered mechanical loads. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2005, 283, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Midgett, M.; López, C.S.; David, L.; Maloyan, A.; Rugonyi, S. Increased Hemodynamic Load in Early Embryonic Stages Alters Endocardial to Mesenchymal Transition. Front. Physiol. 2017, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.M.; McPheeters, M.T.; Wang, Y.T.; Ma, P.; Gu, S.; Strainic, J.; Snyder, C.; Rollins, A.M.; Watanabe, M.; Jenkins, M.W. Increased regurgitant flow causes endocardial cushion defects in an avian embryonic model of congenital heart disease. Congenit. Heart Dis. 2017, 12, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, F.J.; Ingber, D.E. Mechanotransduction: All signals point to cytoskeleton, matrix, and integrins. Sci. STKE 2002, 119, pe6. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Corbit, K.C.; Sirerol-Piquer, M.S.; Ramaswami, G.; Otto, E.A.; Noriega, T.R.; Seol, A.D.; Robinson, J.F.; Bennett, C.L.; Josifova, D.J.; et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011, 43, 7767–7784. [Google Scholar] [CrossRef] [PubMed]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Emmer, B.T.; Maric, D.; Engman, D.M. Molecular mechanisms of protein and lipid targeting to ciliary membranes. J. Cell Sci. 2010, 123, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Maimari, N.; Pedrigi, R.M.; Russo, A.; Broda, K.; Krams, R. Integration of flow studies for robust selection of mechanoresponsive genes. Thromb. Haemost. 2016, 115, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Slough, J.; Cooney, L.; Brueckner, M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev. Dyn. 2008, 237, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Hierck, B.P.; Van der Heiden, K.; Alkemade, F.E.; Van de Pas, S.; Van Thienen, J.V.; Groenendijk, B.C.; Bax, W.H.; Van der Laarse, A.; DeRuiter, M.C.; Horrevoets, A.J.; et al. Primary cilia sensitize endothelial cells for fluid shear stress. Dev. Dyn. 2008, 237, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Van der Heiden, K.; Egorova, A.D.; Poelmann, R.E.; Wentzel, J.J.; Hierck, B.P. Role for primary cilia as flow detectors in the cardiovascular system. Int. Rev. Cell Mol. Biol. 2011, 290, 87–119. [Google Scholar] [CrossRef] [PubMed]
- Van der Heiden, K.; Hierck, B.P.; Krams, R.; de Crom, R.; Cheng, C.; Baiker, M.; Pourquie, M.J.; Alkemade, F.E.; DeRuiter, M.C.; Gittenberger-de Groot, A.C.; et al. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis 2008, 196, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Egorova, A.D.; Khedoe, P.P.; Goumans, M.J.; Yoder, B.K.; Nauli, S.M.; Ten Dijke, P.; Poelmann, R.E.; Hierck, B.P. Lack of primary cilia primes shear-induced endothelial-to-mesenchymal transition. Circ. Res. 2011, 108, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Hogers, B.; DeRuiter, M.C.; Gittenberger-de Groot, A.C.; Poelmann, R.E. Extraembryonic venous obstructions lead to cardiovascular malformations and can be embryolethal. Cardiovasc. Res. 1999, 41, 87–99. [Google Scholar] [CrossRef]
- Carmeliet, P.; Collen, D. Molecular basis of angiogenesis. Role of VEGF and VE-cadherin. Ann. N. Y. Acad. Sci. 2000, 902, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, N.M.; Caolo, V.; Molin, D.G. Cellular decisions in cardiac outflow tract and coronary development: An act by VEGF and NOTCH. Differentiation 2012, 84, 62–78. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Camenisch, T.D.; Itin, A.; Fishman, G.I.; McDonald, J.A.; Carmeliet, P.; Keshet, E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 2001, 128, 1531–1538. [Google Scholar] [PubMed]
- Smedts, H.P.; Isaacs, A.; de Costa, D.; Uitterlinden, A.G.; van Duijn, C.M.; Gittenberger-de Groot, A.C.; Helbing, W.A.; Steegers, E.A.; Steegers-Theunissen, P. VEGF polymorphisms are associated with endocardial cushion defects: A family-based case-control study. Pediatr. Res. 2010, 67, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, N.M.; Molin, D.G.; Peters, P.P.; Maas, S.; Wisse, L.J.; Van Brempt, R.; Van Munsteren, C.J.; Bartelings, M.M.; Poelmann, R.E.; Carmeliet, P.; et al. Tetralogy of Fallot and alterations in vascular endothelial growth factor-A signaling and notch signaling in mouse embryos solely expressing the VEGF120 isoform. Circ. Res. 2007, 100, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Jahnsen, E.D.; Trindade, A.; Zaun, H.C.; Lehoux, S.; Duarte, A.; Jones, E.A. Notch1 is pan-endothelial at the onset of flow and regulated by flow. PLoS ONE 2015, 10, e0122622. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, G.; Luxán, G.; del Monte-Nieto, G.; Martínez-Poveda, B.; Torroja, C.; Walter, W.; Bochter, M.S.; Benedito, R.; Cole, S.; Martinez, F.; et al. Sequential Notch activation regulates ventricular chamber development. Nat. Cell Biol. 2016, 18, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Engleka, K.A.; Rentschler, S.L.; Manderfield, L.J.; Li, L.; Yuan, L.; Epstein, J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Investig. 2011, 121, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.C.; Laney, A.O.; Smith, R.; Gerfen, J.; Morrissette, J.J.; Woyciechowski, S.; Garbarini, J.; Loomes, K.M.; Krantz, I.D.; Urban, Z.; et al. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum. Mutat. 2010, 31, 594–601. [Google Scholar] [CrossRef] [PubMed]
- McCright, B.; Lozier, J.; Gridley, T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 2002, 129, 1075–1082. [Google Scholar] [PubMed]
- Schneider, L.; Clement, C.A.; Teilmann, S.C.; Pazour, G.J.; Hoffmann, E.K.; Satir, P.; Christensen, S.T. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Gaetano, C.; Antonini, A.; Pompilio, G.; Bracco, E.; Rönnstrand, L.; Heldin, C.H.; Capogrossi, M.C. Different effects of high and low shear stress on platelet-derived growth factor isoform release by endothelial cells: Consequences for smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Bleyl, S.B.; Saijoh, Y.; Bax, N.A.; Gittenberger-de Groot, A.C.; Wisse, L.J.; Chapman, S.C.; Hunter, J.; Shiratori, H.; Hamada, H.; Yamada, S.; et al. Dysregulation of the PDGFRA gene causes inflow tract anomalies including TAPVR: Integrating evidence from human genetics and model organisms. Hum. Mol. Genet. 2010, 19, 1286–1301. [Google Scholar] [CrossRef] [PubMed]
- Bax, N.A.; Lie-Venema, H.; Vicente-Steijn, R.; Bleyl, S.B.; Van Den Akker, N.M.; Maas, S.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Platelet-derived growth factor is involved in the differentiation of second heart field-derived cardiac structures in chicken embryos. Dev. Dyn. 2009, 238, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Dekker, R.J.; Van Soest, S.; Fontijn, R.D.; Salamanca, S.; De Groot, P.G.; VanBavel, E.; Pannekoek, H.; Horrevoets, A.J. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 2002, 100, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Lin, Z.; Jain, M.K. “Go with the flow”: How Krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid. Redox Signal. 2011, 15, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Leyen, T.A.; Fontijn, R.D.; Fledderus, J.O.; Baggen, J.M.; Volger, O.L.; van Nieuw Amerongen, G.P.; Horrevoets, A.J. KLF2-induced actin shear fibers control both alignment to flow and JNK signaling in vascular endothelium. Blood 2010, 115, 2533–2542. [Google Scholar] [CrossRef] [PubMed]
- Fontijn, R.D.; Volger, O.L.; Van der Pouw-Kraan, T.C.; Doddaballapur, A.; Leyen, T.; Baggen, J.M.; Boon, R.; Horrevoets, A.J. Expression of Nitric Oxide-Transporting Aquaporin-1 Is Controlled by KLF2 and Marks Non-Activated Endothelium In Vivo. PLoS ONE 2015, 10, e0145777. [Google Scholar] [CrossRef] [PubMed]
- Chiplunkar, A.R.; Lung, T.K.; Alhashem, Y.; Koppenhaver, B.A.; Salloum, F.N.; Kukreja, R.C.; Haar, J.L.; Lloyd, J.A. Krüppel-like factor 2 is required for normal mouse cardiac development. PLoS ONE, 2013, 8, e54891. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yu, Q.; Shin, J.T.; Sebzda, E.; Bertozzi, C.; Chen, M.; Mericko, P.; Stadtfeld, M.; Zhou, D.; Cheng, L.; et al. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev. Cell 2006, 11, 845–857. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.L.; Parnall, M.; Loughna, S. Effect of altered haemodynamics on the developing mitral valve in chick embryonic heart. J. Mol. Cell. Cardiol. 2017, 108, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Vozzi, F.; Bianchi, F.; Ahluwalia, A.; Domenici, C. Hydrostatic pressure and shear stress affect endothelin-1 and nitric oxide release by endothelial cells in bioreactors. Biotechnol. J. 2014, 9, 146–154. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Hayes, E.M.; Lehoux, S.; Jeremy, J.Y.; Horrevoets, A.J.; Newby, A.C. Characterization of the differential response of endothelial cells exposed to normal and elevated laminar shear stress. J. Cell. Physiol. 2011, 226, 2841–2848. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.X.; Cai, S.X.; Wang, P.Q.; Ouyang, K.Q.; Wang, Y.L.; Xu, S.R. Shear-induced changes in endothelin-1 secretion of microvascular endothelial cells. Microvasc. Res. 2002, 63, 209–217. [Google Scholar] [CrossRef]
- Groenendijk, B.C.; Stekelenburg-de Vos, S.; Vennemann, P.; Wladimiroff, J.W.; Nieuwstadt, F.T.; Lindken, R.; Westerweel, J.; Hierck, B.P.; Ursem, N.T.; Poelmann, R.E. The endothelin-1 pathway and the development of cardiovascular defects in the haemodynamically challenged chicken embryo. J. Vasc. Res. 2008, 45, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Kurihara, H.; Oda, H.; Maemura, K.; Nagai, R.; Ishikawa, T.; Yazaki, Y. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J. Clin. Investig. 1995, 96, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Silacci, P.; Harrison, V.J.; Hayoz, D. Nitric oxide synthase expression in endothelial cells exposed to mechanical forces. Hypertension 1998, 32, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Balligand, J.L.; Feron, O.; Dessy, C. eNOS activation by physical forces: From short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol. Rev. 2009, 89, 481–534. [Google Scholar] [CrossRef] [PubMed]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig. 1998, 101, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Song, W.; Lu, X.; Hamilton, J.A.; Lei, M.; Peng, T.; Yee, S.P. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation 2002, 106, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lu, X.; Xiang, F.L.; Poelmann, R.E.; Gittenberger-de Groot, A.C.; Robbins, J.; Feng, Q. Nitric oxide synthase-3 deficiency results in hypoplastic coronary arteries and postnatal myocardial infarction. Eur. Heart J. 2014, 35, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Bartelings, M.M.; Goumans, M.J.; DeRuiter, M.C.; Poelmann, R.E. The arterial and cardiac epicardium in development, disease and repair. Differentiation 2012, 84, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Cooley, J.R.; Yatskievych, T.A.; Antin, P.B. Embryonic expression of the transforming growth factor beta ligand and receptor genes in chicken. Dev. Dyn. 2014, 243, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Azhar, M.; Schultz, J.; Grupp, I.; Dorn, G.W., 2nd; Meneton, P.; Molin, D.G.; Gittenberger-de Groot, A.C.; Doetschman, T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003, 14, 391–407. [Google Scholar] [CrossRef]
- Doetschman, T.; Barnett, J.V.; Runyan, R.B.; Camenisch, T.D.; Heimar, R.L.; Granzier, H.L.; Conway, S.J.; Azhar, M. Transforming growth factor beta signaling in adult cardiovascular diseases and repair. Cell Tissue Res. 2012, 347, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, B.P.; Duim, S.N.; Moerkamp, A.T.; Goumans, M.J. TGFβ and BMP signaling in cardiac cushion formation: Lessons from mice and chicken. Differentiation 2012, 84, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Papoutsi, T.; Luna-Zurita, L.; Prados, B.; Zaffran, S.; De la Pompa, J.L. Bmp2 and Notch cooperate to pattern the embryonic endocardium. Development 2018, 145, dev163378. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, B.; Ito, Y.; Makita, T.; Sasaki, T.; Chai, Y.; Sucov, H.M. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev. Biol. 2006, 289, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Walshe, T.E.; De la Paz, N.G.; D’Amore, P.A. The role of shear-induced transforming growth factor-β signaling in the endothelium. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2608–2617. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bohanan, C.S.; Neumann, J.C.; Lingrel, J.B. KLF2 transcription factor modulates blood vessel maturation through smooth muscle cell migration. J. Biol. Chem. 2008, 283, 3942–3950. [Google Scholar] [CrossRef] [PubMed]
- Dekker, R.J.; Boon, R.A.; Rondaij, M.G.; Kragt, A.; Volger, O.L.; Elderkamp, Y.W.; Meijers, J.C.; Voorberg, J.; Pannekoek, H.; Horrevoets, A.J. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 2006, 107, 4354–4363. [Google Scholar] [CrossRef] [PubMed]
- Rushton, D.I. Pathology of placenta. In Textbook of Fetal and Perinatal Pathology; Wigglesworth, J.S., Singer, D.B., Eds.; Blackwell Scientific Publications: Boston, MA, USA, 1991; pp. 161–220. [Google Scholar]
- Rosenthal, G.L. Patterns of prenatal growth among infants with cardiovascular malformations: Possible fetal hemodynamic effects. Am. J. Epidemiol. 1996, 143, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Groenenberg, I.A.; Wladimiroff, J.W.; Hop, W.C. Fetal cardiac and peripheral arterial flow velocity waveforms in intrauterine growth retardation. Circulation 1989, 80, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Jantzen, D.W.; Moon-Grady, A.J.; Morris, S.A.; Armstrong, A.K.; Berg, C.; Dangel, J.; Fifer, C.G.; Frommelt, M.; Gembruch, U.; Herberg, U.; et al. Hypoplastic Left Heart Syndrome with Intact or Restrictive Atrial Septum: A Report From the International Fetal Cardiac Intervention Registry. Circulation 2017, 136, 1346–1349. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hamburger Hamilton (HH) Stages | 8–20 | 22–23 | 24 | Total 18–24 | ||||
|---|---|---|---|---|---|---|---|---|
| Number of Embryos | n = 19 | % | n = 16 | % | n = 28 | % | n = 61 | % |
| Normal | 0 | 0 | 0 | 0 | 77 | 25 | 7 | 11 |
| Disturbed looping | 6 | 32 | 5 | 31 | 9 | 32 | 20 | 32 |
| Hypoplastic atrioventricular (AV) cushions | 7 | 37 | 8 | 50 | 9 | 32 | 24 | 38 |
| Hypoplastic outflow tract (OFT) cushions | 3 | 16 | 2 | 13 | 6 | 21 | 11 | 17 |
| Myocardium left atrium | 8 | 42 | 4 | 25 | 0 | 0 | 16 | 25 |
| Myocardium ventricle | 3 | 16 | 3 | 19 | 6 | 21 | 12 | 19 |
| Pharyngeal arch arteries | 1 | 5 | 1 | 6 | 7 | 25 | 9 | 14 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poelmann, R.E.; Gittenberger-de Groot, A.C. Hemodynamics in Cardiac Development. J. Cardiovasc. Dev. Dis. 2018, 5, 54. https://doi.org/10.3390/jcdd5040054
Poelmann RE, Gittenberger-de Groot AC. Hemodynamics in Cardiac Development. Journal of Cardiovascular Development and Disease. 2018; 5(4):54. https://doi.org/10.3390/jcdd5040054
Chicago/Turabian StylePoelmann, Robert E., and Adriana C. Gittenberger-de Groot. 2018. "Hemodynamics in Cardiac Development" Journal of Cardiovascular Development and Disease 5, no. 4: 54. https://doi.org/10.3390/jcdd5040054
APA StylePoelmann, R. E., & Gittenberger-de Groot, A. C. (2018). Hemodynamics in Cardiac Development. Journal of Cardiovascular Development and Disease, 5(4), 54. https://doi.org/10.3390/jcdd5040054