
Self-Healable and Super-Tough Double-Network Hydrogel Fibers from Dynamic Acylhydrazone Bonding and Supramolecular Interactions

1
Institute of Textiles and Clothing, The Hong Kong Polytechnic University, Hong Kong 999077, China
2
Key Laboratory of Functional Polymer Materials, College of Chemistry, Nankai University, Tianjin 300071, China
*
Author to whom correspondence should be addressed.
Gels 2022, 8(2), 101; https://doi.org/10.3390/gels8020101
Submission received: 31 December 2021
/
Revised: 27 January 2022
/
Accepted: 1 February 2022
/
Published: 8 February 2022
(This article belongs to the Special Issue Gels Horizons: From Science to Smart Materials)
Abstract
:Macroscopic hydrogel fibers are highly desirable for smart textiles, but the fabrication of self-healable and super-tough covalent/physical double-network hydrogels is rarely reported. Herein, copolymers containing ketone groups were synthesized and prepared into a dynamic covalent hydrogel via acylhydrazone chemistry. Double-network hydrogels were constructed via the dynamic covalent crosslinking of copolymers and the supramolecular interactions of iota-carrageenan. Tensile tests on double-network and parental hydrogels revealed the successful construction of strong and tough hydrogels. The double-network hydrogel precursor was wet spun to obtain macroscopic fibers with controlled drawing ratios. The resultant fibers reached a high strength of 1.35 MPa or a large toughness of 1.22 MJ/m3. Highly efficient self-healing performances were observed in hydrogel fibers and their bulk specimens. Through the simultaneous healing of covalent and supramolecular networks under acidic and heated conditions, fibers achieved rapid and near-complete healing with 96% efficiency. Such self-healable and super-tough hydrogel fibers were applied as shape memory fibers for repetitive actuating in response to water, indicating their potential in intelligent fabrics.
1. Introduction
Hydrogels are soft materials with a crosslinked network structure. Macroscopic fibers developed from functional hydrogels are ideal candidates for smart textiles (e.g., fibrous actuators [1], hygiene textiles [2] and flexible sensors [3,4,5]). However, hydrogel fibers have undesirable mechanical weaknesses and brittleness, which have limited their real uses, as has the complexity of processing flowable dope into crosslinked gel strands. In our previous work, hydrogels with high toughness have been synthesized and applied in cell cultures [6], shape memory actuators [7,8,9] and intelligent textiles [2,10]. Based on dynamic covalent bonding and the self-assembly of polysaccharide networks, self-healable hydrogel fibers with mechanical robustness would be attractive for the further development of hydrogel textiles.
Improvements on their mechanical properties have been focused on the hydrogel area. Recent strategies include mechanochemical self-strengthening (e.g., self-growing [11], strain-induced crystallization [12] and stored length releasing [13]), the introduction of sacrificial structures (e.g., interpenetrating network [9,14] and supramolecular interaction [15,16]) and the prevention of crack propagation (e.g., nanocomposites [17,18,19] and macromolecular microsphere composites [20,21]). Among these strategies, double-network (DN) structures have been the most successful strategy to construct strong and tough hydrogels since the pioneering work was conducted in 2003 [22]. By synthesizing stiff and ductile networks simultaneously or sequentially, energy could be dissipated in such hydrogels during mechanical deformation to avoid destructive stress concentration on polymer strands. Based on polymerized covalent networks and physically crosslinked polysaccharide networks, hybrid DN hydrogels have exhibited ultra-high stretchability and toughness that makes them suitable for fiber spinning [8,23,24]. However, the irreversible destruction of covalent networks in such hydrogels is gradually intensified during cyclic deformation and has led to fatigue fracture [25]. To avoid the embrittling caused by fractured polymer strands, the development of a self-healable network is essential for durable and reliable hydrogel fibers.
Self-healable networks rely on the reversibility of crosslinks. Physical networks in DN hydrogels are usually crosslinked via supramolecular interactions, which were widely found to be self-repairable. Typical examples include hydrogen bonding-induced helix bundle assembly (e.g., carrageenan [9], agar [26], gelatin [27] and DNA [28]), electrostatic attraction (e.g., negatively charged groups and multivalent cations [23], poly ionic liquids and negatively charged crosslinkers [29]) and aggregation domains (e.g., semi-crystallinity [30] and hydrophobic interaction [31]). In DN hydrogels, the major challenge is the recovery of damaged covalent networks. Dynamic covalent crosslinks are desirable to achieve complete healing in DN structures, which involves the exchanging of the reaction partner or the formation of new dynamic covalent bonds. The regeneration of dynamic covalent bonds usually requires a trigger from external stimuli. Recently reported examples include the disulfide exchange reaction (redox-responsive) [32,33], imine condensation (pH-responsive) [34,35] and Diels–Alder chemistry (heat-responsive) [36]. By incorporating functional components to achieve the conversion of specific stimuli to other types (e.g., turning acoustic energy to heat), more self-healing hydrogels that are desirable for industry could be developed and applied in future.
The spinning of hydrogel has been challenging primarily due to the weakness of the nascent fiber and the difficulty of processing crosslinked networks where polymer chains are not flowable. The representative routes for constructing hydrogel fibers are ‘monomers to network’ and ‘macromolecules to network’. To overcome the low viscosity of monomer solutions, which leads to the breakage of nascent fiber, a precursor (containing monomers, initiators and crosslinkers) usually polymerizes in tubular templates, which are lubricated or super-hydrophobic [37]. Although fibrous hydrogels with millimeter levels of thickness (thin fibers are poorly demoldable) have been fabricated by such reactive spinning methods, its industrialization has encountered challenges to achieving high-speed manufacturing and obtaining fibers at the micrometer level [38]. In comparison, the more scalable spinning route in the textile industry is by constructing networks using macromolecules. As a facile method to obtain macroscopic hydrogel fibers, wet-spinning has utilized rapid physical interactions to solidify injected dope columns in coagulation baths (the use of covalent crosslinks has rarely been reported in recent literature [39]). Typical solidification strategies include constructing electrostatic attraction (e.g., -COO− and multivalent cations [40] and -NH3+ and negatively charged groups [41]), hydrogen bonding (e.g., agarose double helixes [42] and collagen nanofibrils [43]) and aggregation (e.g., destroying colloidal stability [44] and dehydration by non-solvents [42]). The introduction of multiple and synergistic interactions to several biopolymers (e.g., carrageenan [45], agar [46] and gelatin [30]) offers toughening for nascent fibers. Via the simultaneous or sequential synthesis of covalent networks in physical crosslinked nascent fibers, DN hydrogel fibers are able to be fabricated and find wide applications [47].
In this study, acylhydrazone bonding was utilized for synthesizing a dynamic covalent hydrogel. By introducing a supramolecular network, self-healable DN hydrogels could be constructed with significantly enhanced tensile properties in contrast with their single network (SN) parents.
2. Results and Discussion
2.1. SN Hydrogels from Dynamic Covalent and Supramolecular Networks
Prior to the preparation of DN hydrogels, the parental polyacrylamide-co-diacetone acrylamide (PAD) and iota-carrageenan (IC) single-network hydrogels were synthesized and studied. In order to construct dynamic covalent networks, PAD copolymers were synthesized via radical polymerization (reaction in Figure S1). At a temperature of 70 °C, acrylamide (AM) and diacetone acrylamide (DAAM) monomers were initiated by ammonium persulfate (APS), and randomly copolymerized into macromolecular chains. To provide the successful copolymerization, PAD-5 and parallelly synthesized polyacrylamide (PAM) were selected as typical examples for Fourier transform infrared spectroscopy (FTIR) (Figure 1a) and 1H-NMR (b) characterizations. The FTIR spectrum of PAD-5 shows a peak at 1740 cm−1 (absent in the PAM), which was assigned to ketone carbonyl groups (-CH2-CO-CH3). The 1H-NMR spectrum of PAD-5 showed new signals at 1.28 and 2.79–3.31 ppm, in contrast with that of PAM. This evidence proves the successful copolymerization of DAAM monomers. By calculating the integral area in the NMR curve (peak 1 = 33.5, peak 5 = 224.0), the molar percentage of DAAM was calculated as 4.98% (equal to peak1 divided by peak5 three times), which is close to the feeding ratio (5.00%). This small difference is attributed to the similar reactivity of AM and DAAM in radical polymerization [48]. Furthermore, the weight average molecule weight (Mw) and polydispersity index (PDI) of the copolymers were determined via gel permeation chromatography (GPC) (Table S1). Although the Mw of PAD copolymers slightly increased with the growth of the DAAM feeding ratios (DAAM is larger than AM), the Mw and PDI values of all copolymers are close to PAM due to the same reaction temperature and time. Based on the approximately equal chain length, the comparison of PAD hydrogels was allowed for the selection of a suitable spinning dope component.
Covalent SN hydrogels were synthesized using the keto-hydrazide reaction. The evidence of crosslinking is presented in Figure S2a. After adding adipic dihydrazide (ADH) into the aqueous solution of PAD-5, gelation was observed whether at reacting temperatures of 25 or 70 °C, and the crosslinking was accelerated by higher temperatures. Meanwhile, no gelation occurred in the parallelly incubated PAM and ADH mixture solution (Figure S2b). This phenomenon indicates that copolymer chains were crosslinked via a reaction between DAAM units and ADH. Specifically, ketone groups on the DAAM units reacted with the hydrazide groups on the ADH molecules to form acylhydrazone bonds (the reaction is diagramed in Figure S3). This bonding presented sensitivity to pH, since the adding of formic acid (which adjusted the pH to 1) and the neutralization by triethylamine (which adjusted the pH to 6) led to the decomposition and regeneration of PAD hydrogel, respectively (Figure 2a). The formation and breakage of acylhydrazone bonds were controlled by the pH (critical value = 2.5 [49]) and was allowed to be repeated (Figure 2c).
The gelation time is crucial for the spinning of hydrogels fiber; thus, the gelation process of PAD SN hydrogels at different temperatures was monitored through dynamic time sweep rheological experiments. As presented in Figure 3a, the storage modulus (G’) increased with the reaction time, and the growth trend at 70 °C is significantly higher than that at 25 °C. Additionally, the increasing of the phase shift angle followed the same trend (Figure 3b), indicating that the PAD precursor transferred from a liquid into a solid more rapidly at a higher temperature. The gelation times were recorded where crossovers of the storage modulus and loss modulus (phase shift angle = 45 degree) appeared. At a higher temperature, the gelation time shifted earlier (215 min at 25 °C and 20 min at 70 °C) due to the acceleration of the keto-hydrazide reaction. The kinetics of gelation were accelerated approximately ten times faster at 70 °C, indicating the faster equilibrium of acylhydrazone formation and transamination when the motions of PAD segments and ADH became intensified [50]. Such rapid equilibrium kinetics is desirable in the self-healing of damaged networks, where the rapid repairing of cracks by reformed acylhydrazone linkages is allowed [51]. Therefore, the gelation time of PAD SN hydrogel is controllable by manipulating the reaction temperature, in which 70 °C was chosen for further rapid and in situ hydrogel fiber preparation.
Physical SN hydrogel was prepared by cooling down IC aqueous solution. IC is a typical thermo-sensitive polysaccharide. As demonstrated in Figure 2b, the IC solution crosslinked into hydrogel when the temperature dropped below 65 °C, while the hydrogel decomposed into the solution when the temperature rose above 65 °C. This sol-gel transition is attributed to the thermo-sensitivity of IC (Figure 2d) [8]. When the temperature dropped below the upper critical solution temperature (UCST), the IC chains self-assembled to double-helix bundles via the hydrogen bonds. Then, the negatively charged sulfonate groups on the IC bundles and chains associated with cations via electrostatic attraction. These two supramolecular interactions caused the gelation of IC and were able to offer sacrificial bonds in the double-network structure. When the temperature rose above the UCST, the physical interactions disassociated and led to the decomposition of hydrogel.
Tensile tests were performed on PAD SN hydrogels to nominate a suitable copolymer in further studies. As presented in Figure 4a, PAD SN hydrogels presented higher strength but lower stretchability with the increase in DAAM contents, which decided the crosslinking density in these hydrogels. In comparison with PAD-1 (too weak) and PAD-10 (too brittle) hydrogels, PAD-5 hydrogel was nominated for further preparation of DN hydrogel due to its great toughness.
2.2. Tensile Properties and Self-Healing Performances of DN Hydrogels
To assess the feasibility of spinning, PAD-5/IC DN hydrogels were synthesized and compared with their parent SN hydrogels. As illustrated in Figure 4b, PAD-5 SN hydrogels presented a stretchable (ε~300%) but weak (σ < 0.1 MPa) performance, while IC SN hydrogels showed a stiff (E~1 MPa) but brittle (ε~50%) behavior, with an obvious yielding point. This yielding performance is attributed to the destruction of electrostatic attraction between the sulfonate groups and calcium ions [52]. The PAD-5/IC DN hydrogels exhibited significantly higher strength and stretchability due to the energy dissipation of sacrificed IC physical network. Moreover, PAD-5/IC hydrogels presented increased modulus at a highly stretched state, indicating the successful transition of the brittle facture to a ductile type [23]. At the highly stretched stage, polymer strands were aligned, which allowed the formation of highly oriented conformation during spinning.
Since both parental SN hydrogels presented stimuli-responsive sol-gel transitions, the PAD-5/IC DN hydrogel should be self-healable. Given that the sequential healing of two networks is time consuming, simultaneous healing is preferred in real applications. As demonstrated in Figure 4c, two differently colored DN hydrogel rectangles were joint together after cutting. By smearing formic acid on the interface and incubating at a temperature above the UCST of the IC, the joint segments completely healed into one rectangle, which was able to be stretched (Figure 4d) [53]. The self-healing process at the interface between two segments was recorded by optical microscopy (Figure S4a–c). With the increase in the healing time, the gap width between two segments decreased from 400 μm initially to 100 μm at 2 h. The disappearing of the crack was observed at 4 h of healing time, which indicated the near-complete self-healing of hydrogel segments. The spread of dye molecules between two segments led to a purple color at the boundary after healing, hinting the exchange of materials (including reaction partners) at the interface [50].
2.3. Spinning, Tensile Properties, Self-Healing Efficiencies and Shape Memory of DN Hydrogel Fibers
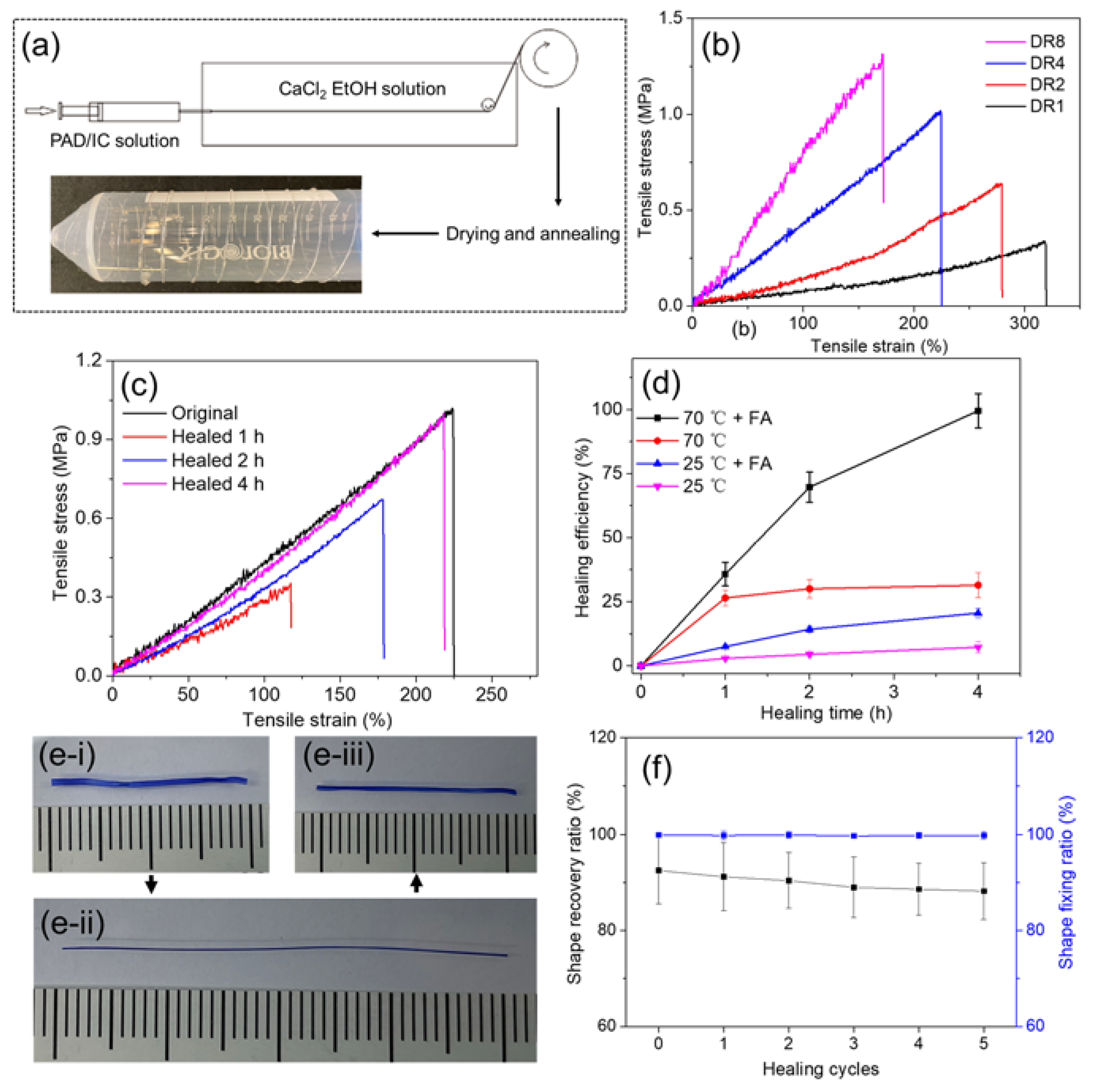
Based on the PAD-5/IC hydrogel, DN fibers could be fabricated by wet spinning. A stable and flowable spinning dope is essential for continuous spinning. As a result, the pH of PAD-5/IC hydrogel precursor was adjusted to 1 in order to prevent covalent crosslinking before spinning, while the temperature of the syringe was retained at 70 °C to avoid physical gelation. To reduce the effect of the acidic hydrolysis of IC, the spinning dope was immediately used after it was obtained. The wet-spinning process is demonstrated in Figure 5a. Under the flowable state, the spinning dope was injected into the coagulation bath (10 wt% of CaCl2 ethanol solution). Through the electrostatic attraction between the calcium divalent cations and the negatively charged sulfonate groups on the IC [54], the dope column rapidly crosslinked into the nascent fiber and was progressively strengthened by the coagulation bath due to the continuous penetration of cations and the dehydration from the ethanol. During the spinning, the pH of the nascent fiber was neutralized via the diffusion of protons. Although the majority of the ADH was not reacted with the PAD, the loss of the crosslinker in the coagulation bath was negligible due to the dissolvability of the ADH in ethanol. After being collected on roller, the fibers were dried at 70 °C to remove the residual formic acid and annealed for higher crystallinity and dimensional stability.
During spinning, nascent fibers were stretched for a higher orientation degree. The influence of drawing ratios (DR) on the tensile properties and morphology was investigated. As presented in Figure 5b and summarized in Table 1, fibers with increased DR became stronger and stiffer, although the stretchability was decreased. These changes evidently indicated that the polymer strands in the highly stretched fibers became more aligned along the longitude direction, which boosted the fiber strength. The cross-section images of the fibers also showed that the fibers became thinner with the increase in the DR (Figure S5). Additionally, the swelling capacity of fibers was slightly affected by the aligned structure, which led to the restricted expansion of the network along the longitudinal direction as it absorbed water [55]. The least swelled fiber reached more than 10 times the swelling degree at the equilibrium state, which already meets the requirement of biomedical applications [14,56]. The toughness of the PAD-5/IC-DR4 fiber reached an attractively high value of 1.22 MJ/m3, which is suitable for applications in real products.
Since the self-healing performance was observed in the PAD-5/IC bulk hydrogels, the healing efficiency of the toughest DR4 fiber was quantitively evaluated by tensile tests (Figure 5d). After immersing the breaking ends of the failed fiber into formic acid and attaching the two ends together, the healing sample was sealed and incubated at 70 °C for 1, 2 and 4 h. In comparison with the original fiber, the healing fiber showed gradually enhanced strength and modulus with increasing time. The 4 h healed fiber reached 1.03 MPa of strength, which almost equaled that of the original fiber (giving it a 96% healing efficiency). Although the covalent and physical networks healed simultaneously, their respective contribution to the healing was studied by healing under different conditions (Figure 5d). At 70 °C, with formic acid smeared on interface, the fiber achieved highly efficient and rapid healing. The healed fiber at 25 °C without formic acid showed almost no healing (<15%). Without raising the temperature, the supramolecular network was not healed, but the dynamic regeneration of the acylhydrazone bonds contributed approximately 20% of healing at 4 h. Similarly, without the assistance of formic acid, although the fiber rapidly healed its supramolecular network in 1 h, the invalidation of the covalent network led to a low efficiency of <30% at 4 h. Benefiting from the convenient processing of fiber healing, such self-healable and super-tough DN hydrogel fibers may be applied in healable fabrics and flexible actuators.
As a potential shape memory fiber (SMF) with self-healing properties for actuating uses, the PAD-5/IC-DR4 fibers were swelled (to reach 90% of water content, Figure 5e-i) and post-drawn to 100% of strain. By drying under room conditions, the stretched shape of the fibers were fixed without obvious contraction after releasing the stretching (Figure 5e-ii). When the fixed SMFs were rehydrated in water, their glass transition temperature kept decreasing with the increase in water content. Water acted as plasticizer and led to the shape recovery of the SMFs (Figure 5e-iii), which started contracting in the longitudinal direction when their glass transition temperature dropped below the water temperature. Furthermore, by cutting the recovered fibers and healing them under optimal conditions (70 °C with acid), the shape recovery processes were repeated for five cycles. The shape fixing and recovery ratios of SMFs were determined in Figure 5f. With number of healing cycles increased from zero to five, the shape fixing ratios remained the same level of almost 100%. The shape recovery ratios kept decreasing from 92.5% (virgin SMFs) to 88.2% (healed five times), indicating the good repeatability and fatigue resistance of highly healed SMFs during multiple healing and recovering processes [57].
3. Conclusions
In summary, PAD copolymers with different monomer molar ratios were successfully synthesized via radical polymerization. The structure of PAD-5 was characterized by FTIR, NMR and GPC. The ketone groups on the DAAM allowed the formation of dynamic acylhydrazone bonds with the ADH, which led to the gelation of PAD SN hydrogels. The gelation time at 70 °C was determined through time sweep experiment evaluation. The IC SN hydrogels were prepared by facile cooling down. The PAD and IC hydrogels presented reversible self-healing performance in response to pH and temperature, respectively. Tensile studies on the PAD, IC and PAD/IC hydrogels indicated the construction of DN hydrogels and a nominated composition of spinning dope. The simultaneous healing of both networks in the PAD-5/IC hydrogel was achieved by heating and formic acid smearing on the interface. Via facile wet spinning, PAD-5/IC hydrogel fibers with DR ranging from 1 to 8 were fabricated. Such fibers reached a high strength of 1.35 MPa or a high toughness of 1.22 MJ/m3. The effects of healing time, temperature and formic acid on the healing efficiencies were studied with a tensile test. DR4 fiber was able to achieve highly efficient and rapid healing to reach 96% of its original strength in 4 h. The PAD-5/IC fibers were applied as SMFs for actuating in response to water. The highly efficient self-healing property of SMFs allowed the shape memory process to be repeated five times, indicating that these super-tough and self-healable DN hydrogel fibers may be ideal candidates in smart textiles.
4. Materials and Methods
4.1. Materials
AM (>99.9%), IC (>99%, 4.48 wt% K+ and 2.68 wt% Ca2+, tested via inductively coupled plasma mass spectrometry), calcium chloride (>99%) and APS (>99%) were purchased from Sigma-Aldrich Co (Shanghai, China). DAAM (>98%) and ADH (>99%) were purchased from Tokyo Chemical Industry Co., Ltd. (Shanghai, China). All chemicals were used as received without purification.
4.2. Synthesis of PAM and PAD
AM, DAAM and APS (0.31 mol% of monomers) were sequentially dissolved in degassed water at 70 °C. After 8 h of stirring (70 °C, 150 rpm, N2 degassing), copolymers were precipitated with acetone (equivalent mass of obtained mixture) and vacuum dried (50 °C, 24 h) (yield: 98.3%). The copolymers were denoted as PAD-n, where n stands for the molar percentage of DAAM. The unit contents of PAD-5 were evaluated by a 1H-NMR; 1H-NMR (400 MHz, D2O, r.t.) δ 1.28 (s, 6H, (CH3)2C(NH-)CH2-), 1.34–1.83 (br, 2H, -CH2- of the main ethylene chain), 1.91–2.36 (br, 1H, -CH- in the main ethylene chain), 2.14 (s, 3H, CH3-CO), 2.79–3.31 (br, 2H, CH3-CO-CH2-). PAM was parallelly synthesized with an equivalent monomer molar concentration (yield: 97.8%). The Mw and PDI (equal to Mw/Mn) of the synthesized polymers were determined via GPC (water mobile phase, reference: polyethylene oxide, 1260 ALS, Aglient, Santa Clara, USA).
4.3. Preparation of PAD, IC and PAD/IC Hydrogels
PAD hydrogel precursors were obtained by dissolving ADH (75 mol% of DAAM) in PAD aqueous solutions and rapidly injecting the mixture into a Teflon model (incubated at 25 or 70 °C for 8 h) to prepare covalent SN hydrogels. The time sweep experiments (frequency = 1 Hz, strain = 1.0%) were parallelly performed using a rheometer (MCR702, Anton Paar Inc,. Graz, Austria) with a flat plate of 25 mm diameter and a gap of 1 mm to evaluate the gelation time of the PAD-5 hydrogel. After the precursor solution was transferred onto the parallel plate (silicone oil was used to avoid water evaporation during the test) and reached the predetermined temperature, the G′, G″ and phase shift angle as functions of time were recorded. The gelation time was recorded when G′ = G″. The IC hydrogel precursor was obtained by dissolving IC and CaCl2 (75 mol% of sulfate groups) in water at 70 °C and rapidly injecting the mixture into a Teflon model until cooling down to room temperature. The PAD/IC DN hydrogel precursor was prepared after nominating PAD-5 as an optimized copolymer for further study. PAD-5 and IC precursors were mixed to obtain a homogeneous solution (PAD:IC = 3:1 w/w), and then rapidly injected into a Teflon model (incubated at 70 °C for 8 h). The water contents of all hydrogel specimens were fixed at 92.0 wt%.
4.4. Spinning of PAD/IC Hydrogel Fibers
PAD/IC hydrogel fibers were fabricated using a wet-spinning method. The homogeneous spinning dope was obtained by dissolving ADH (75 mol% of DAAM) in the degassed and acidic (pH = 1, adjusted by formic acid) aqueous solution that contained PAD (6.0 wt%) and IC (2.0 wt%). The dope (maintained at 70 °C) was injected into a coagulation bath (10.0 wt% of CaCl2 ethanol solution, 30 cm long, room temperature) using a syringe pump (spinning speed = 0.36 m/min, 18 G of spinneret, LSP01-1A, Baoding Longer Precision Pump Co., Ltd., Baoding, China). The solidified nascent fibers were collected on a drum with different winding rates and were monitored by a contact tachometer (DT-2236, Lutron Electronic Enterprise Co., Ltd., Taipei, China). The drawing ratios (DR, winding rate versus spinning speed) of nascent fibers were controlled as 1, 2, 4 and 8. All nascent fibers were oven dried (70 °C, 8 h) and annealed.
4.5. Fabrication and Shape Recovery Study of PAD/IC SMFs
Swelled PAD-5/IC-DR4 hydrogel fibers (to reach 90% of water content) were stretched to 100% of strain and air dried under stretching to obtain shape-fixed SMFs (fixed strain = εm). SMFs were immersed into water at 20 °C to observe shape recovery performance. The residual strain (εr) of recovered fibers was determined when recovery was completed. The shape fixing ratio was calculated by equation: Rf = εm/stretched strain × 100%. Shape recovery ratios were calculated by equation: Rr = (εm − εr)/εm × 100%. Recovered fibers were cut and healed (by smearing formic acid at the interface and maintaining them at 70 °C for 4 h). Shape recovery experiments were repeated on healed SMFs for 5 healing cycles to evaluate repetitive shape memory performance.
4.6. Characterizations
The chemical structures were characterized via 1H-NMR spectroscopy (Bruker Avance-III 400 MHz, Zurich, Switzerland). Chemical shifts were referenced to the solvent values (δ = 4.79 ppm for D2O). The chemical structure of each specimen was determined by FTIR (Perkin Elmer spectrum 100, Waltham, Massachusetts, USA) using wavenumbers between 4000 and 650 cm−1 with a 4 cm−1 resolution (spectra between 1630 and 1830 cm−1 were presented). Tensile properties were tested using a uniaxial tensile machine (Instron 4411, Norwood, MA, USA) with a 10.0 N load cell at an extension rate of 50.0 mm/min at 20.0 °C and 65.0% of relative humidity. The starting distance between two clamps was set as 20.0 mm. The tensile strength (σ, kPa) and stretchability (ε, %) were recorded at failure point. Young’s modulus (E, kPa) was calculated by fitting the initial linear region of stress–strain curve (tensile strain = 5~10%). The toughness (Γ, MJ/m3) was calculated according to the integral area of the tensile curves (MPa over mm/mm). The healing efficiency was calculated according to the percentage of the healed sample strength versus the original fiber strength. All tensile hydrogel samples were prepared in Teflon molds with a dumb-bell shape (50.0 × 4.0 × 2.5 mm3). All tensile properties were reported as mean and standard deviation (sample size = 6).
Hydrogel specimens were immersed into DI water until equilibrium swelling, and their weights (We) were then measured. After completely oven drying them, the weights of dry hydrogels (Wd) were recorded. The swelling capacities (Q) were calculated by expression: Q = (We − Wd)/Wd.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/gels8020101/s1, Figure S1: Scheme of radical polymerization of PAD; Figure S2: (a) Synthesis of PAD hydrogel; (i) PAD aqueous solution; (ii) crosslinked PAD SN hydrogel. (b) No gelation occurred in PAM solution; (iii) PAM aqueous solution; (iv) PAM and ADH mixture solution (scale bar = 1 cm). Figure S3: Crosslinking reaction between PAD chains and ADH. Figure S4: Interface cross-section images of PAD-5/IC hydrogel segments with healing time of (a) 0, (b) 2 and (c) 4 h (dye stained: above by rhodamine B, below by methylene blue). Figure S5: Cross-section images of (a) DR1, (b) DR2, (c) DR4 and (d) DR8 PAD-5/IC fibers. Table S1: Molecular weight (Mw) and polydispersity of synthesized polymers determined by GPC.
Author Contributions
Conceptualization, J.H. and B.F.; methodology, J.H.; software, J.H. and C.L.; validation, J.H.; formal analysis, J.H.; investigation, J.H.; resources, J.H. and C.L.; data curation, J.H. and C.L.; writing—original draft preparation, J.H.; writing—review and editing, J.H., B.F. and Z.L.; visualization, J.H. and C.L.; supervision, B.F.; project administration, B.F.; funding acquisition, B.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Hong Kong Research Grant Council GRF 15215617.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, J.R.; Wang, J.F.; Chen, Z.; Fang, S.L.; Zhu, Y.; Baughman, R.H.; Jiang, L. Tunable, Fast, Robust Hydrogel Actuators Based on Evaporation-Programmed Heterogeneous Structures. Chem. Mater. 2017, 29, 9793–9801. [Google Scholar] [CrossRef]
- Wang, X.W.; Hu, H.W.; Wang, W.Y.; Lee, K.I.; Gao, C.; He, L.; Wang, Y.F.; Lai, C.L.; Fei, B.; Xin, J.H. Antibacterial modification of an injectable, biodegradable, non-cytotoxic block copolymer-based physical gel with body temperature-stimulated sol-gel transition and controlled drug release. Colloids Surf. B Biointerfaces 2016, 143, 342–351. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, Y.; Liu, H.; Wang, Z.; Ling, H.; Wang, C.; Ni, J.; Çelebi-Saltik, B.; Wang, X.; Meng, X.; et al. Room-Temperature-Formed PEDOT:PSS Hydrogels Enable Injectable, Soft, and Healable Organic Bioelectronics. Adv. Mater. 2020, 32, 1904752. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, X.; Jiang, N.; Yetisen, A.K.; Yuk, H.; Yang, C.; Khademhosseini, A.; Zhao, X.; Yun, S.-H. Highly Stretchable, Strain Sensing Hydrogel Optical Fibers. Adv. Mater. 2016, 28, 10244–10249. [Google Scholar] [CrossRef]
- Yetisen, A.K.; Jiang, N.; Fallahi, A.; Montelongo, Y.; Ruiz-Esparza, G.U.; Tamayol, A.; Zhang, Y.S.; Mahmood, I.; Yang, S.A.; Kim, K.S.; et al. Glucose-Sensitive Hydrogel Optical Fibers Functionalized with Phenylboronic Acid. Adv. Mater. 2017, 29, 1606380. [Google Scholar] [CrossRef]
- Hua, J.C.; Li, Z.; Xia, W.; Yang, N.; Gong, J.X.; Zhang, J.F.; Qiao, C.S. Preparation and properties of EDC/NHS mediated crosslinking poly (gamma-glutamic acid)/epsilon-polylysine hydrogels. Mater. Sci. Eng. C 2016, 61, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Chen, L.; Li, Y.L.; Lee, K.I.; Fei, B. Super-tough hydrogels from shape-memory polyurethane with wide-adjustable mechanical properties. J. Mater. Sci. 2017, 52, 4421–4434. [Google Scholar] [CrossRef]
- Lu, X.K.; Chan, C.Y.; Lee, K.I.; Ng, P.F.; Fei, B.; Xin, J.H.; Fu, J. Super-tough and thermo-healable hydrogel—Promising for shape-memory absorbent fiber. J. Mater. Chem. B 2014, 2, 7631–7638. [Google Scholar] [CrossRef]
- Hua, J.; Liu, C.; Ng, P.F.; Fei, B. Bacterial cellulose reinforced double-network hydrogels for shape memory strand. Carbohydr. Polym. 2021, 259, 117737. [Google Scholar] [CrossRef]
- Wang, X.W.; Hu, H.W.; Yang, Z.Y.; He, L.; Kong, Y.Y.; Fei, B.; Xin, J.H. Smart hydrogel-functionalized textile system with moisture management property for skin application. Smart Mater. Struct. 2014, 23, 125027. [Google Scholar] [CrossRef]
- Matsuda, T.; Kawakami, R.; Namba, R.; Nakajima, T.; Gong, J.P. Mechanoresponsive self-growing hydrogels inspired by muscle training. Science 2019, 363, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Morimoto, N.; Jiang, L.; Kawahara, S.; Noritomi, T.; Yokoyama, H.; Mayumi, K.; Ito, K. Tough hydrogels with rapid self-reinforcement. Science 2021, 372, 1078–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zheng, X.; Ouchi, T.; Kouznetsova, T.B.; Beech, H.K.; Av-Ron, S.; Matsuda, T.; Bowser, B.H.; Wang, S.; Johnson, J.A.; et al. Toughening hydrogels through force-triggered chemical reactions that lengthen polymer strands. Science 2021, 374, 193–196. [Google Scholar] [CrossRef]
- Hua, J.C.; Fei, B. Super-Tough Polyacrylamide/iota-Carrageenan Double-Network Hydrogels Strengthened By Bacterial Cellulose Microclusters. Mater. Today Proc. 2019, 16, 1497–1501. [Google Scholar] [CrossRef]
- Bilici, C.; Ide, S.; Okay, O. Yielding Behavior of Tough Semicrystalline Hydrogels. Macromolecules 2017, 50, 3647–3654. [Google Scholar] [CrossRef]
- Chen, Y.; Pang, X.H.; Dong, C.M. Dual Stimuli-Responsive Supramolecular Polypeptide-Based Hydrogel and Reverse Micellar Hydrogel Mediated by Host-Guest Chemistry. Adv. Funct. Mater. 2010, 20, 579–586. [Google Scholar] [CrossRef]
- Wang, J.F.; Lin, L.; Cheng, Q.F.; Jiang, L. A Strong Bio-Inspired Layered PNIPAM-Clay Nanocomposite Hydrogel. Angew. Chem. Int. Ed. 2012, 51, 4676–4680. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.H.; Hai, Z.B.; Cui, C.H.; Li, H.H.; Chen, J.F.; Yu, S.H. In Situ Controlled Synthesis of Thermosensitive Poly(N-isopropylacrylamide)/Au Nanocomposite Hydrogels by Gamma Radiation for Catalytic Application. Small 2012, 8, 930–936. [Google Scholar] [CrossRef]
- Jaiswal, M.K.; Xavier, J.R.; Carrow, J.K.; Desai, P.; Alge, D.; Gaharwar, A.K. Mechanically Stiff Nanocomposite Hydrogels at Ultralow Nanoparticle Content. ACS Nano 2016, 10, 246–256. [Google Scholar] [CrossRef]
- Huang, T.; Xu, H.G.; Jiao, K.X.; Zhu, L.P.; Brown, H.R.; Wang, H.L. A novel hydrogel with high mechanical strength: A macromolecular microsphere composite hydrogel. Adv. Mater. 2007, 19, 1622–1626. [Google Scholar] [CrossRef]
- Zhao, J.; Jiao, K.; Yang, J.; He, C.; Wang, H. Mechanically strong and thermosensitive macromolecular microsphere composite poly(N-isopropylacrylamide) hydrogels. Polymer 2013, 54, 1596–1602. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Sun, J.Y.; Zhao, X.H.; Illeperuma, W.R.K.; Chaudhuri, O.; Oh, K.H.; Mooney, D.J.; Vlassak, J.J.; Suo, Z.G. Highly stretchable and tough hydrogels. Nature 2012, 489, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wei, D.; Chen, H.; Zhu, L.; Jiao, C.; Liu, G.; Huang, L.; Yang, J.; Wang, L.; Zheng, J. Simultaneous Enhancement of Stiffness and Toughness in Hybrid Double-Network Hydrogels via the First, Physically Linked Network. Macromolecules 2015, 48, 8003–8010. [Google Scholar] [CrossRef]
- Kim, J.; Zhang, G.; Shi, M.; Suo, Z. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science 2021, 374, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, Q.; Hu, R.D.; Wang, H.; Newby, B.M.Z.; Chang, Y.; Zheng, J. Mechanically strong hybrid double network hydrogels with antifouling properties. J. Mater. Chem. B 2015, 3, 5426–5435. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Wei, K.C.; Zhang, K.Y.; Yang, B.G.; Tian, F.; Wang, G.X.; Bian, L.M. One-pot solvent exchange preparation of non-swellable, thermoplastic, stretchable and adhesive supramolecular hydrogels based on dual synergistic physical crosslinking. NPG Asia Mater. 2018, 10, e455. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.H.; Yao, C.; Kou, X.H.; Tang, J.P.; Luo, D.; Yang, D.Y. A Fluorescent Biofunctional DNA Hydrogel Prepared by Enzymatic Polymerization. Adv. Healthc. Mater. 2018, 7, 7. [Google Scholar] [CrossRef]
- Ren, Y.; Guo, J.; Liu, Z.; Sun, Z.; Wu, Y.; Liu, L.; Yan, F. Ionic liquid-based click-ionogels. Sci. Adv. 2019, 5, eaax0648. [Google Scholar] [CrossRef] [Green Version]
- He, Q.Y.; Huang, Y.; Wang, S.Y. Hofmeister Effect-Assisted One Step Fabrication of Ductile and Strong Gelatin Hydrogels. Adv. Funct. Mater. 2018, 28, 1705069. [Google Scholar] [CrossRef]
- Taylor, D.L.; Panhuis, M.I.H. Self-Healing Hydrogels. Adv. Mater. 2016, 28, 9060–9093. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.T.; Mredha, M.T.I.; Na, J.Y.; Seon, J.-K.; Cui, J.; Jeon, I. Multifunctional poly (disulfide) hydrogels with extremely fast self-healing ability and degradability. Chem. Eng. J. 2020, 394, 124941. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, W.; Wang, Q.; Xu, C.; Tang, Q.; Yang, H. An injectable, dual responsive, and self-healing hydrogel based on oxidized sodium alginate and hydrazide-modified poly (ethyleneglycol). Molecules 2018, 23, 546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathan, M.; Kovaříček, P.; Jurissek, C.; Senf, A.; Dallmann, A.; Thünemann, A.F.; Hecht, S. Control of imine exchange kinetics with photoswitches to modulate self-healing in polysiloxane networks by light illumination. Angew. Chem. Int. Ed. 2016, 55, 13882–13886. [Google Scholar] [CrossRef]
- Basu, S.; Pacelli, S.; Paul, A. Self-healing DNA-based injectable hydrogels with reversible covalent linkages for controlled drug delivery. Acta Biomater. 2020, 105, 159–169. [Google Scholar] [CrossRef]
- Madl, C.M.; Heilshorn, S.C. Rapid diels-alder cross-linking of cell encapsulating hydrogels. Chem. Mater. 2019, 31, 8035–8043. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, J.; Liao, W.; Yang, Z. Liquid Crystal Elastomer Twist Fibers towards Rotating Microengines. Adv. Mater. 2022, 34, 2107840. [Google Scholar] [CrossRef]
- Wei, P.; Hou, K.; Chen, T.; Chen, G.; Mugaanire, I.T.; Zhu, M. Reactive spinning to achieve nanocomposite gel fibers: From monomer to fiber dynamically with enhanced anisotropy. Mater. Horiz. 2020, 7, 811–819. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Sun, J.; He, H.; Li, B.; Ma, C.; Liu, K.; Zhang, H. Bioinspired and Mechanically Strong Fibers Based on Engineered Non-Spider Chimeric Proteins. Angew. Chem. 2020, 132, 8225–8229. [Google Scholar] [CrossRef]
- Guimarães, C.F.; Ahmed, R.; Mataji-Kojouri, A.; Soto, F.; Wang, J.; Liu, S.; Stoyanova, T.; Marques, A.P.; Reis, R.L.; Demirci, U. Engineering Polysaccharide-Based Hydrogel Photonic Constructs: From Multiscale Detection to the Biofabrication of Living Optical Fibers. Adv. Mater. 2021, 33, 2105361. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, J.; Liu, J.; Wang, Y.; Guo, J.; Zhong, Z.; Liu, X. High mechanical performance based on the alignment of cellulose nanocrystal/chitosan composite filaments through continuous coaxial wet spinning. Cellulose 2021, 28, 7995–8008. [Google Scholar] [CrossRef]
- Bao, X.; Hayashi, K.; Li, Y.; Teramoto, A.; Abe, K. Novel agarose and agar fibers: Fabrication and characterization. Mater. Lett. 2010, 64, 2435–2437. [Google Scholar] [CrossRef]
- Koeck, K.S.; Salehi, S.; Humenik, M.; Scheibel, T. Processing of Continuous Non-Crosslinked Collagen Fibers for Microtissue Formation at the Muscle-Tendon Interface. Adv. Funct. Mater. 2021, 2112238. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, A.; He, X.; Li, Q.; Sun, J.; Lei, Z.; Liu, Z.H. Full-Temperature All-Solid-State Ti3C2Tx/Aramid Fiber Supercapacitor with Optimal Balance of Capacitive Performance and Flexibility. Adv. Funct. Mater. 2021, 31, 2010944. [Google Scholar]
- Ng, P.F.; Hua, J.; Liu, C.; Wang, Y.; Yin, R.; Fei, B. Solar Energy Storage Silks via Coaxial Wet Spinning. ACS Mater. Lett. 2020, 2, 801–807. [Google Scholar] [CrossRef]
- Liu, J.; Xue, Z.; Zhang, W.; Yan, M.; Xia, Y. Preparation and properties of wet-spun agar fibers. Carbohydr. Polym. 2018, 181, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Song, J.C.; Chen, S.; Sun, L.J.; Guo, Y.F.; Zhang, L.Z.; Wang, S.L.; Xuan, H.X.; Guan, Q.B.; You, Z.W. Mechanically and Electronically Robust Transparent Organohydrogel Fibers. Adv. Mater. 2020, 32, 1906994. [Google Scholar] [CrossRef] [PubMed]
- Ido, Y.; Maçon, A.L.; Iguchi, M.; Ozeki, Y.; Koeda, S.; Obata, A.; Kasuga, T.; Mizuno, T. Construction of enzyme-encapsulated fibermats from the cross-linkable copolymers poly(acrylamide)-co-poly(diacetone acrylamide) with the bi-functional cross-linker, adipic acid dihydrazide. Polymer 2017, 132, 342–352. [Google Scholar] [CrossRef]
- Guo, Z.; Ma, W.; Gu, H.; Feng, Y.; He, Z.; Chen, Q.; Mao, X.; Zhang, J.; Zheng, L. pH-Switchable and self-healable hydrogels based on ketone type acylhydrazone dynamic covalent bonds. Soft Matter 2017, 13, 7371–7380. [Google Scholar] [CrossRef]
- Yang, X.; Liu, G.; Peng, L.; Guo, J.; Tao, L.; Yuan, J.; Chang, C.; Wei, Y.; Zhang, L. Highly efficient self-healable and dual responsive cellulose-based hydrogels for controlled release and 3D cell culture. Adv. Funct. Mater. 2017, 27, 1703174. [Google Scholar] [CrossRef]
- Dirksen, A.; Dirksen, S.; Hackeng, T.M.; Dawson, P.E. Nucleophilic catalysis of hydrazone formation and transimination: Implications for dynamic covalent chemistry. J. Am. Chem. Soc. 2006, 128, 15602–15603. [Google Scholar] [CrossRef]
- Liu, S.J.; Li, L. Recoverable and Self-Healing Double Network Hydrogel Based on kappa-Carrageenan. ACS Appl. Mater. Interfaces 2016, 8, 29749–29758. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, J.; Wang, Y.; Shao, C.; Chi, J.; Li, Z.; Wang, X.; Zhao, Y. Photocontrolled healable structural color hydrogels. Small 2019, 15, 1903104. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Xue, Z.; Liu, J.; Yan, M.; Xia, Y.; Wang, B. Preparation of carrageenan fibers with extraction of Chondrus via wet spinning process. Carbohydr. Polym. 2018, 194, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Mredha, M.T.I.; Guo, Y.Z.; Nonoyama, T.; Nakajima, T.; Kurokawa, T.; Gong, J.P. A Facile Method to Fabricate Anisotropic Hydrogels with Perfectly Aligned Hierarchical Fibrous Structures. Adv. Mater. 2018, 30, 1704937. [Google Scholar] [CrossRef]
- Li, Z.; Wang, L.F.; Hua, J.C.; Jia, S.R.; Zhang, J.F.; Liu, H. Production of nano bacterial cellulose from waste water of candied jujube-processing industry using Acetobacter xylinum. Carbohydr. Polym. 2015, 120, 115–119. [Google Scholar] [CrossRef]
- Ming, X.; Yao, L.; Zhu, H.; Zhang, Q.; Zhu, S. Dramatic and Reversible Water-Induced Stiffening Driven by Phase Separation within Polymer Gels. Adv. Funct. Mater. 2022, 32, 2109850. [Google Scholar] [CrossRef]
Figure 1.
(a) FTIR and (b) 1H-NMR (inset image: structure of PAD) spectra of PAD-5 and PAM.
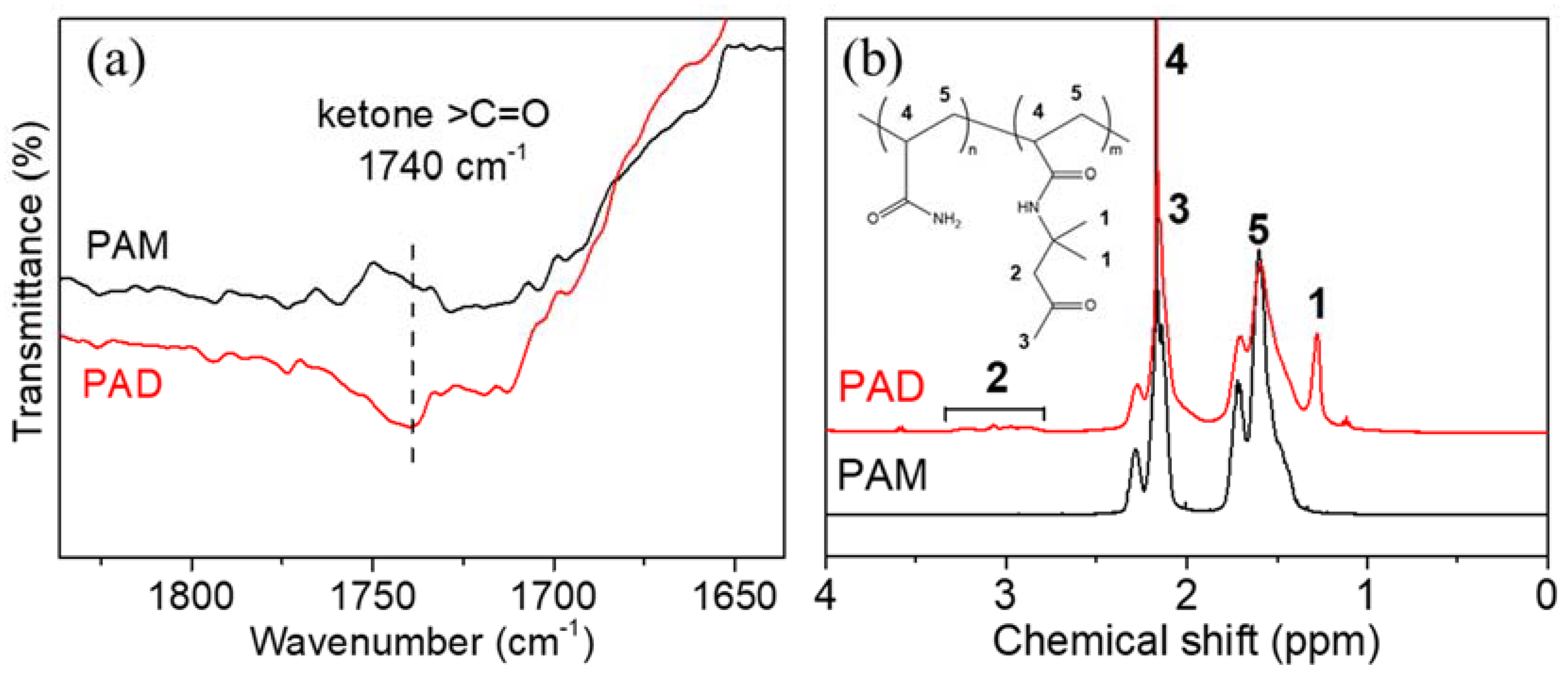
Figure 2.
(a) pH-responsive sol-gel transition of PAD hydrogel: (i) original PAD-5 hydrogel crosslinked by ADH (pH = 6); (ii) hydrogel decomposed under acidic condition (pH = 1); (iii) hydrogel regenerated after pH neutralization (pH = 6); (b) temperature-responsive sol-gel transition of IC hydrogel: (i) IC solution formed above the UCST (upper critical solution temperature); (ii) IC hydrogel formed below the UCST; (c) scheme for pH-responsive sol-gel transition of PAD hydrogel (copolymer chain synthesis is illustrated in the dotted frame); (d) scheme for thermo-responsive sol-gel transition of IC hydrogel (IC structure is illustrated in the dotted frame).
Figure 2.
(a) pH-responsive sol-gel transition of PAD hydrogel: (i) original PAD-5 hydrogel crosslinked by ADH (pH = 6); (ii) hydrogel decomposed under acidic condition (pH = 1); (iii) hydrogel regenerated after pH neutralization (pH = 6); (b) temperature-responsive sol-gel transition of IC hydrogel: (i) IC solution formed above the UCST (upper critical solution temperature); (ii) IC hydrogel formed below the UCST; (c) scheme for pH-responsive sol-gel transition of PAD hydrogel (copolymer chain synthesis is illustrated in the dotted frame); (d) scheme for thermo-responsive sol-gel transition of IC hydrogel (IC structure is illustrated in the dotted frame).
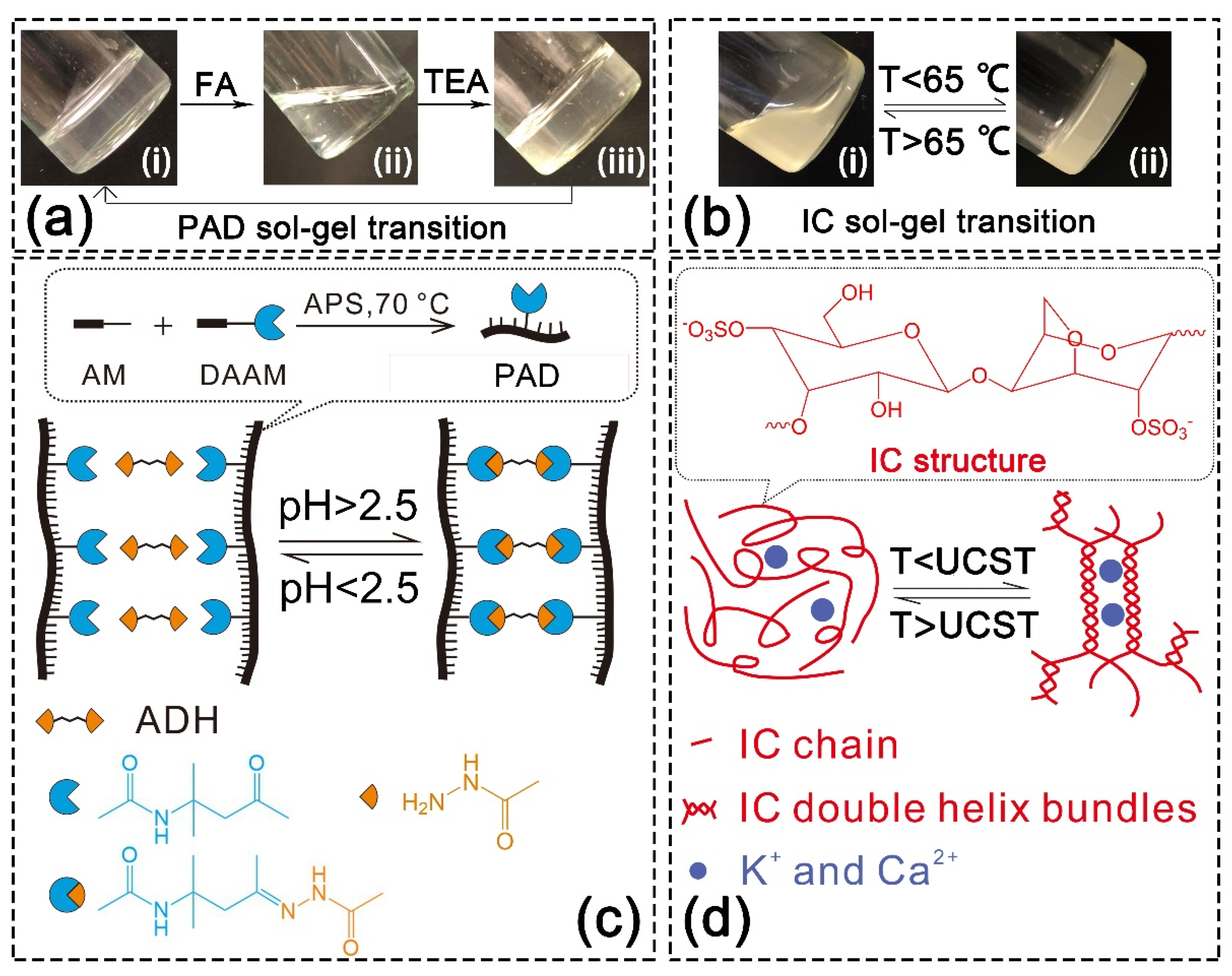
Figure 3.
(a) G′ and G″ and (b) phase shift angle of PAD-5 hydrogel during the gelation process at 25 and 70 °C.
Figure 3.
(a) G′ and G″ and (b) phase shift angle of PAD-5 hydrogel during the gelation process at 25 and 70 °C.

Figure 4.
(a) Typical tensile curves of PAD SN hydrogels; (b) tensile curves of PAD-5, IC SN and PAD-5/IC DN hydrogels; (c) self-healing of PAD-5/IC DN hydrogel segments: rectangle hydrogels stained by (i) methylene blue and (ii) rhodamine B, (iii) self-healed hydrogel segment (interface smeared by formic acid, sealed and incubated at 70 °C for 4 h, all scale bars = 5 mm); (d) healed hydrogel showed stretchability.
Figure 4.
(a) Typical tensile curves of PAD SN hydrogels; (b) tensile curves of PAD-5, IC SN and PAD-5/IC DN hydrogels; (c) self-healing of PAD-5/IC DN hydrogel segments: rectangle hydrogels stained by (i) methylene blue and (ii) rhodamine B, (iii) self-healed hydrogel segment (interface smeared by formic acid, sealed and incubated at 70 °C for 4 h, all scale bars = 5 mm); (d) healed hydrogel showed stretchability.
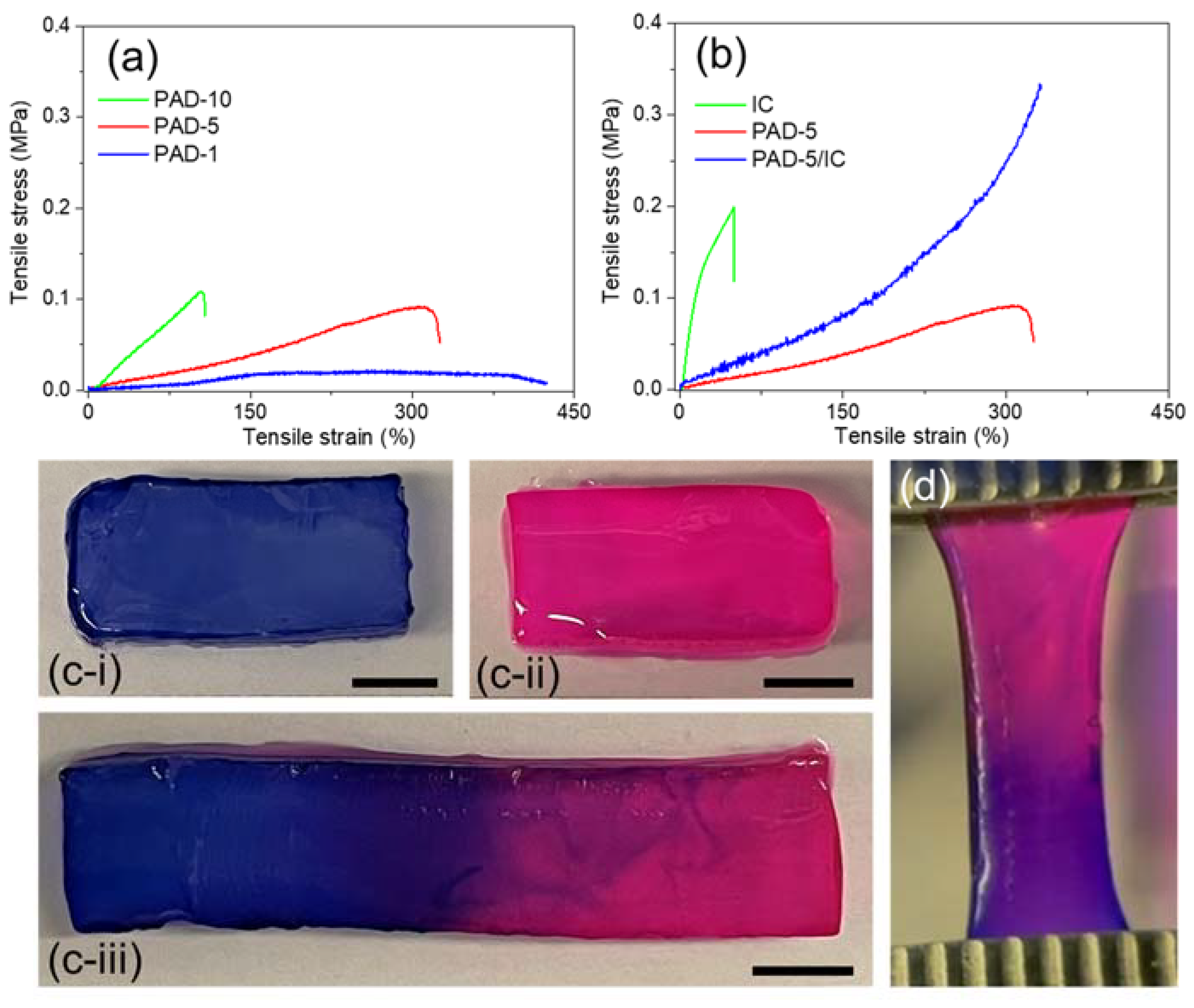
Figure 5.
(a) Scheme of wet-spinning process of PAD-5/IC DN hydrogel fibers (inset photo: DR1 fiber winded on a plastic tube); (b) typical tensile curves of PAD-5/IC DN hydrogel fibers with increased DR; (c) typical tensile curves of PAD-5/IC-DR4 fibers before and after healing for 1, 2 and 4 h; (d) effect of healing time, temperature and formic acid on healing efficiencies; (e) recovery process of PAD-5/IC-DR4 SMF at states of (i) water swelled, (ii) post-drawn and dried, (iii) shape recovered; (f) shape recovery and fixing ratios of PAD-5/IC-DR4 SMF as a function of healing cycles.
Figure 5.
(a) Scheme of wet-spinning process of PAD-5/IC DN hydrogel fibers (inset photo: DR1 fiber winded on a plastic tube); (b) typical tensile curves of PAD-5/IC DN hydrogel fibers with increased DR; (c) typical tensile curves of PAD-5/IC-DR4 fibers before and after healing for 1, 2 and 4 h; (d) effect of healing time, temperature and formic acid on healing efficiencies; (e) recovery process of PAD-5/IC-DR4 SMF at states of (i) water swelled, (ii) post-drawn and dried, (iii) shape recovered; (f) shape recovery and fixing ratios of PAD-5/IC-DR4 SMF as a function of healing cycles.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of tensile properties and swelling capacities of PAD-5/IC hydrogel fibers (standard deviation in bracket, sample size = 6).
Table 1.
Summary of tensile properties and swelling capacities of PAD-5/IC hydrogel fibers (standard deviation in bracket, sample size = 6).
| PAD-5/IC Fibers | Tensile Properties | Swelling Capacity (%) | |||
|---|---|---|---|---|---|
| σ (MPa) | ε (%) | E (MPa) | Γ (MJ/m3) | ||
| DR1 | 0.32 (0.02) | 332.7 (23.99) | 0.11 (0.01) | 0.56 (0.08) | 1235 (11) |
| DR2 | 0.66 (0.09) | 281.6 (16.9) | 0.39 (0.02) | 0.82 (0.09) | 1121 (7) |
| DR4 | 1.07 (0.05) | 225.4 (15.8) | 0.58 (0.04) | 1.22 (0.10) | 1087 (10) |
| DR8 | 1.35 (0.12) | 168.4 (12.5) | 0.81 (0.06) | 1.09 (0.09) | 1030 (9) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hua, J.; Liu, C.; Fei, B.; Liu, Z. Self-Healable and Super-Tough Double-Network Hydrogel Fibers from Dynamic Acylhydrazone Bonding and Supramolecular Interactions. Gels 2022, 8, 101. https://doi.org/10.3390/gels8020101
AMA Style
Hua J, Liu C, Fei B, Liu Z. Self-Healable and Super-Tough Double-Network Hydrogel Fibers from Dynamic Acylhydrazone Bonding and Supramolecular Interactions. Gels. 2022; 8(2):101. https://doi.org/10.3390/gels8020101
Chicago/Turabian StyleHua, Jiachuan, Chang Liu, Bin Fei, and Zunfeng Liu. 2022. "Self-Healable and Super-Tough Double-Network Hydrogel Fibers from Dynamic Acylhydrazone Bonding and Supramolecular Interactions" Gels 8, no. 2: 101. https://doi.org/10.3390/gels8020101
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.