
Chitosan-Silica Hybrid Biomaterials for Bone Tissue Engineering: A Comparative Study of Xerogels and Aerogels

 , , , , ,
, , , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
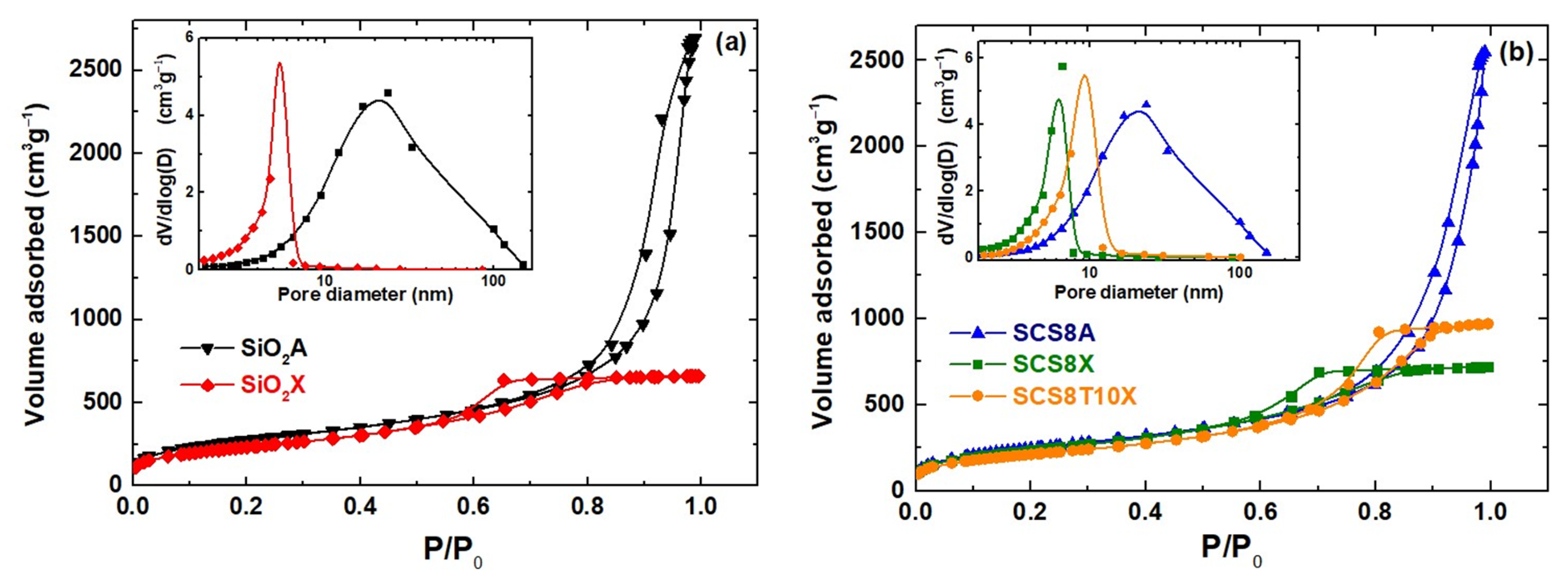
2.1. Physical and Textural Properties of Xerogels and Aerogels
2.2. Thermogravimetric Analysis
2.3. FTIR Spectral Analysis
2.4. In Vitro Bioactivity in SBF
2.5. Osteoblast Behavior
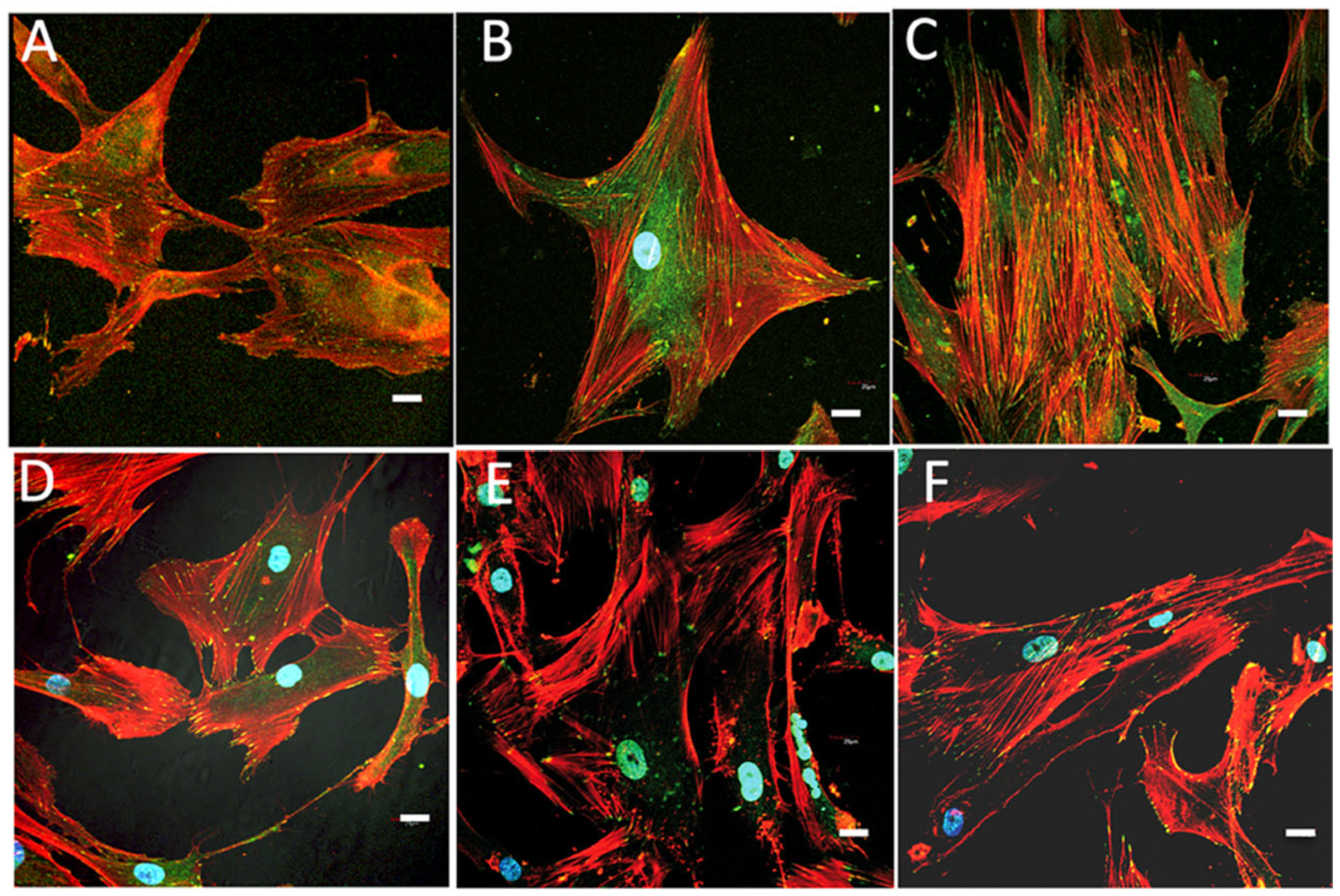
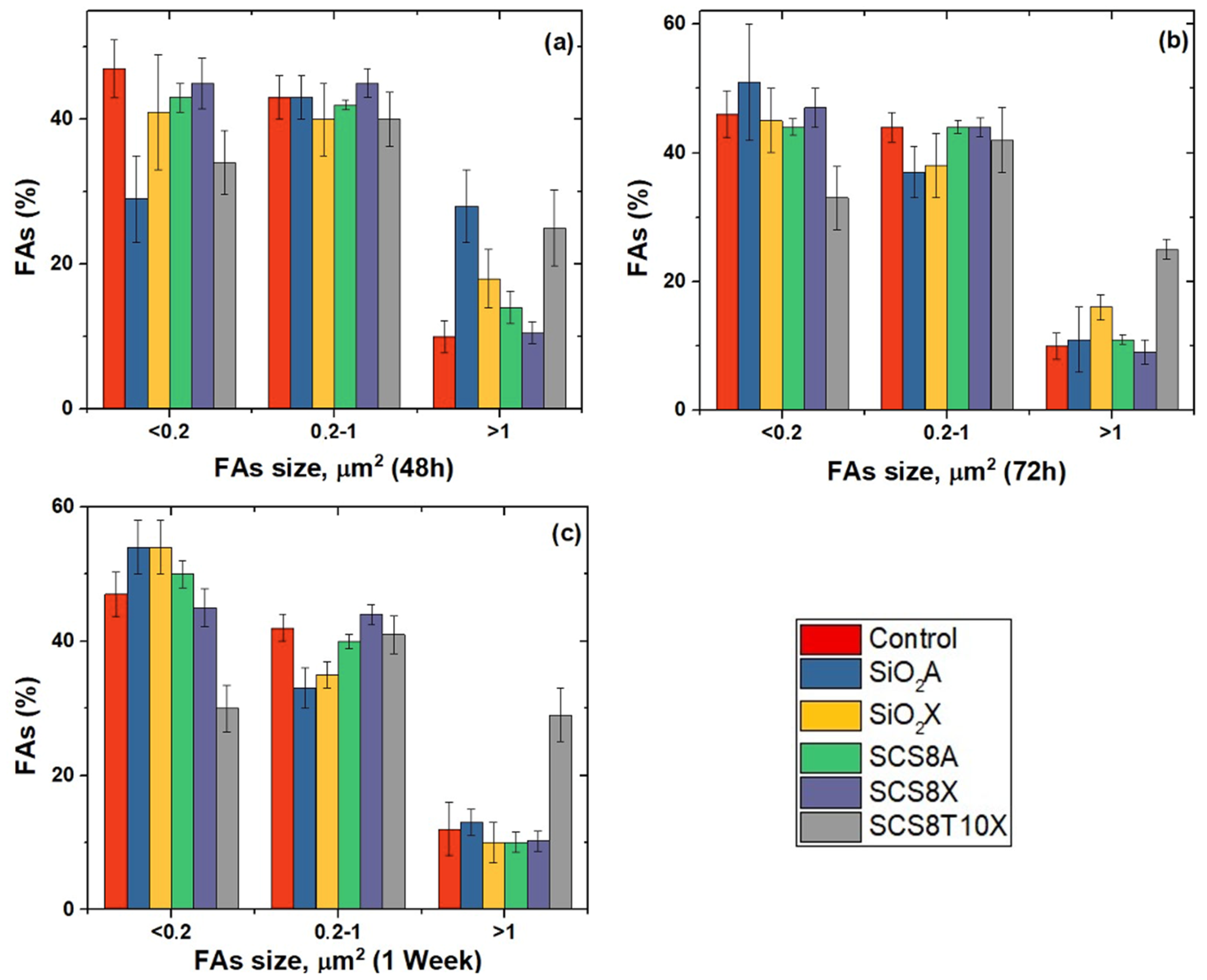
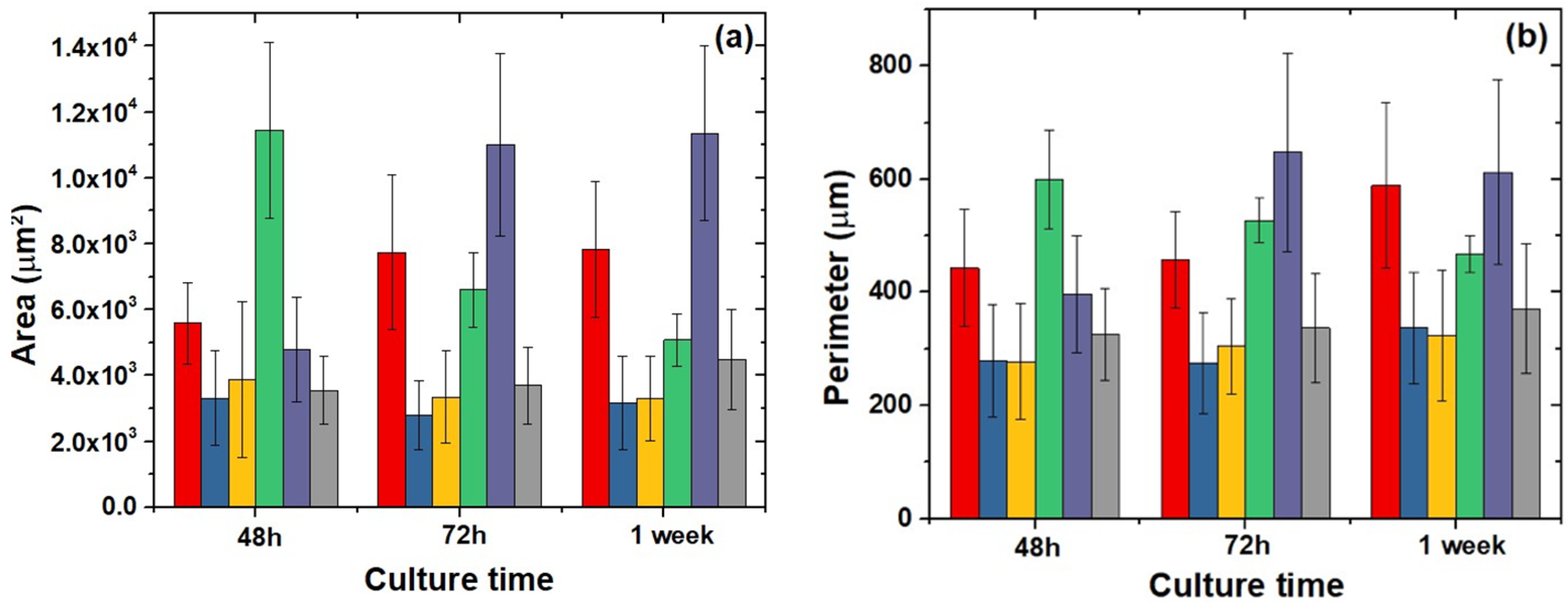
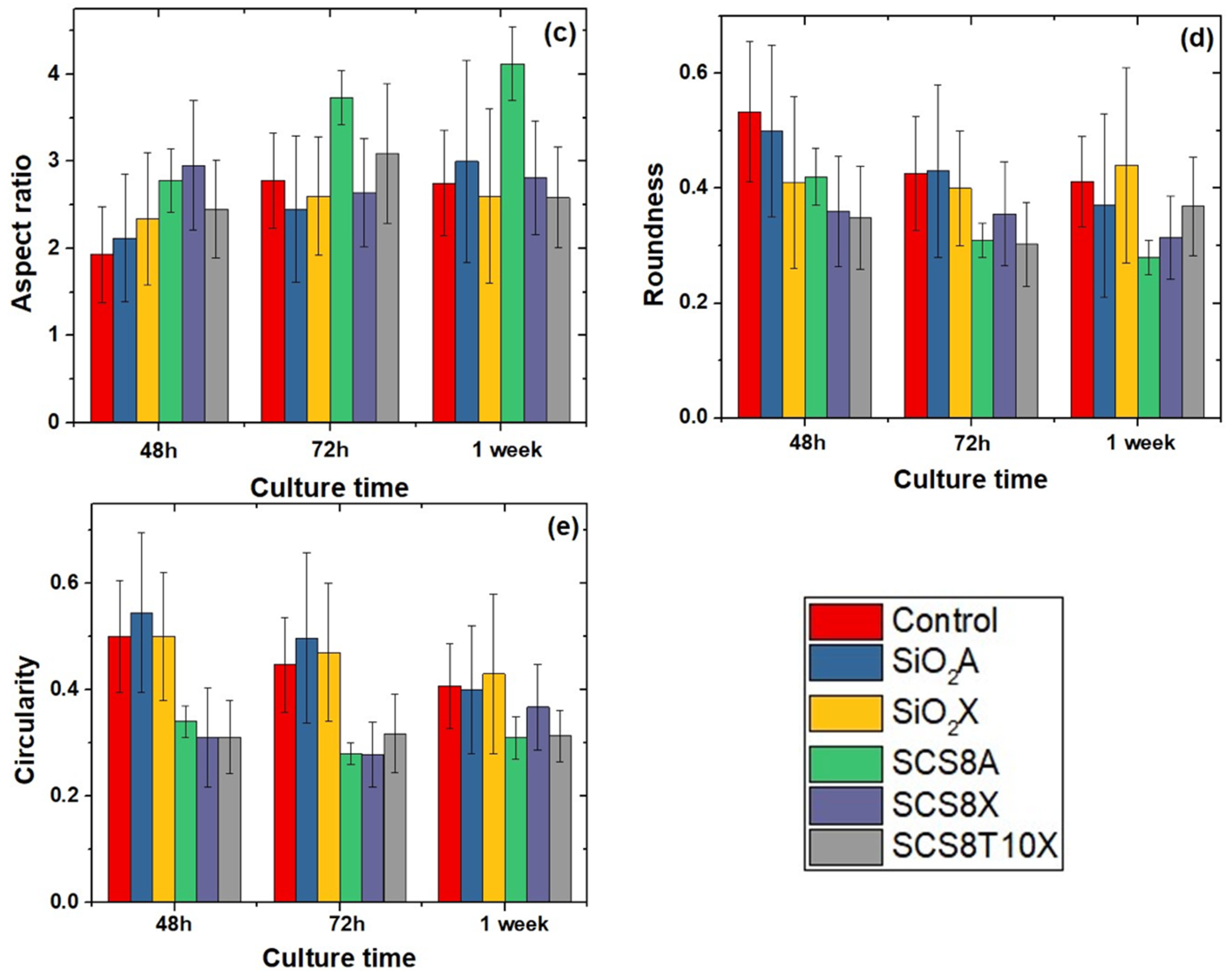
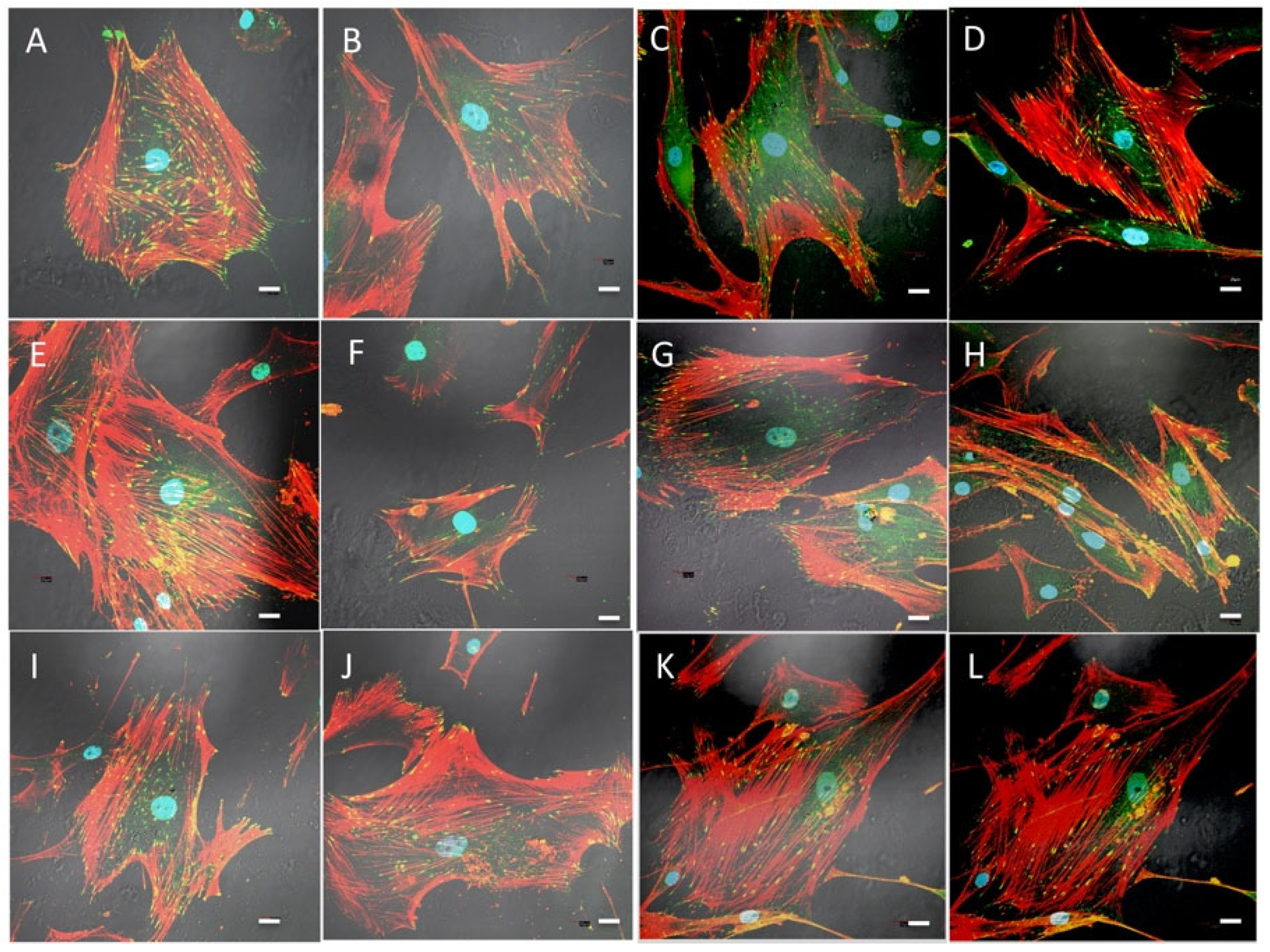
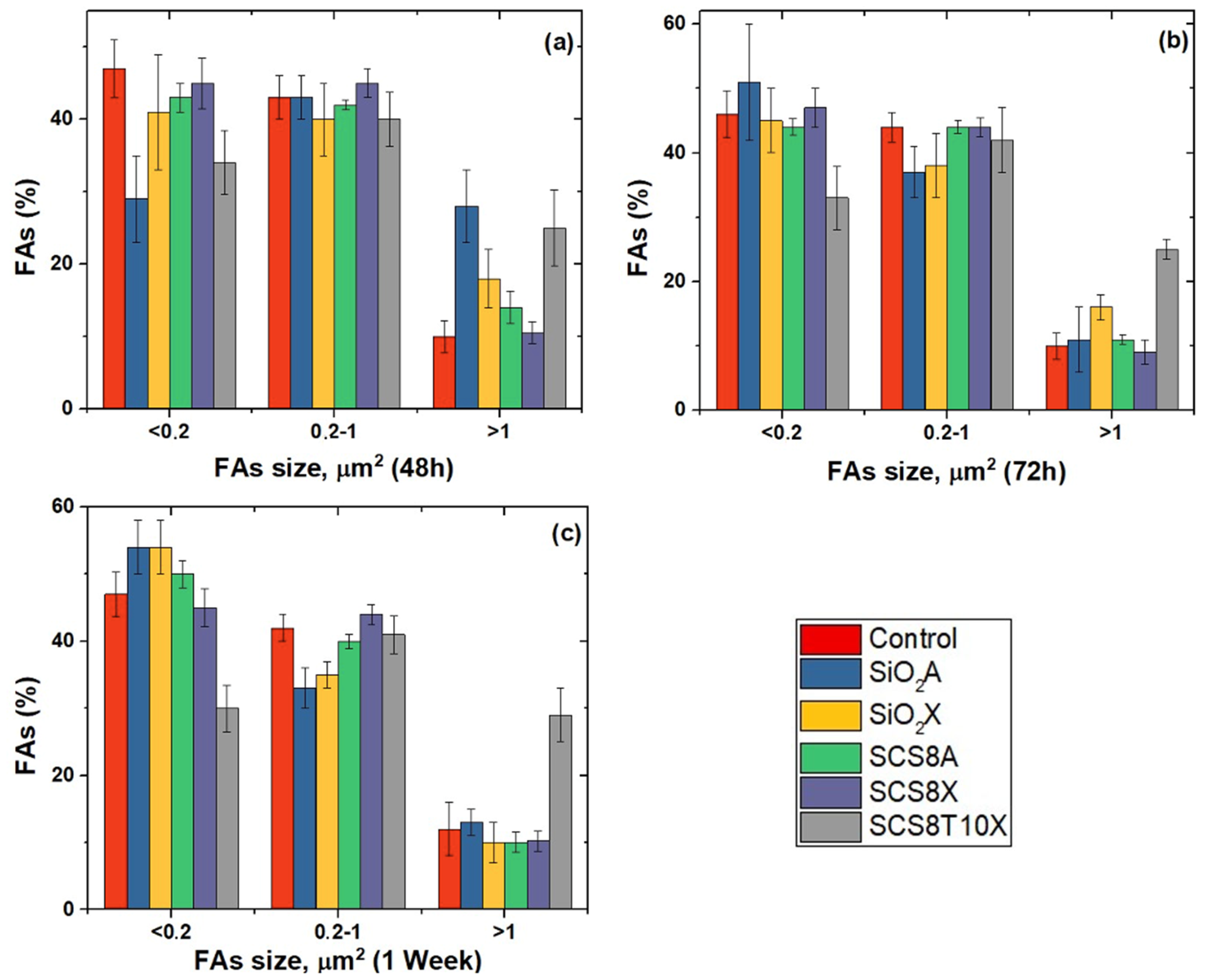
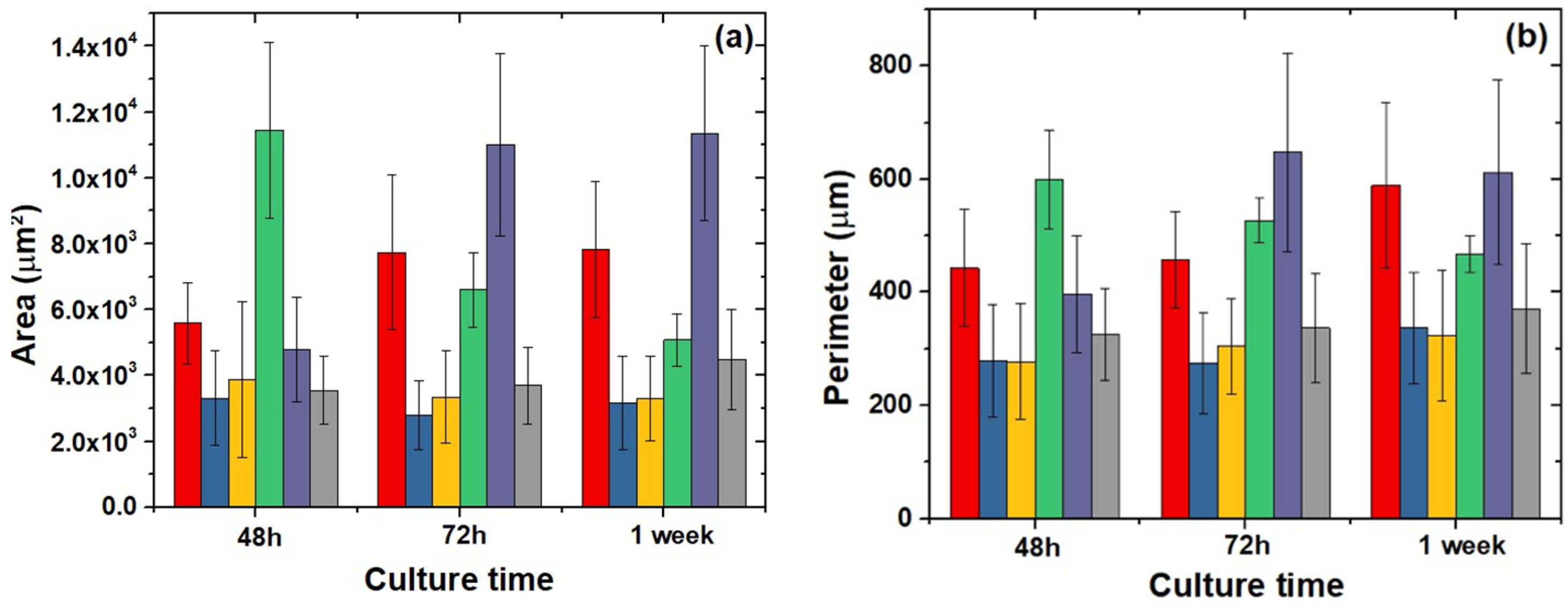
2.6. Cell Morphology, Cytoskeletal Organization, and Focal Adhesions
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Gel Synthesis
4.3. Physical Characterization and Structural Properties
4.4. Thermal Characterization
4.5. Fourier Transform Infrared Spectroscopy
4.6. Evaluation of the Bioactivity in SBF
4.7. Cell Culture
4.8. Live/Dead Cell Assay
4.9. Cell Morphology and Spreading
4.10. Actin Cytoskeletal Organization
4.11. Confocal Examination
4.12. Image Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jeong, J.E.; Park, S.Y.; Shin, J.Y.; Seok, J.M.; Byun, J.H.; Oh, S.H.; Kim, W.D.; Lee, J.H.; Park, W.H.; Park, S.A. 3D Printing of Bone-Mimetic Scaffold Composed of Gelatin/β-Tri-Calcium Phosphate for Bone Tissue Engineering. Macromol. Biosci. 2020, 20, 2000256. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Yague, M.A.; Abbah, S.A.; McNamara, L.; Zeugolis, D.I.; Pandit, A.; Biggs, M.J. Biomimetic approaches in bone tissue engineering: Integrating biological and physicomechanical strategies. Adv. Drug Deliv. Rev. 2015, 84, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Veres, P.; Király, G.; Nagy, G.; Lázár, I.; Fábián, I.; Kalmár, J. Biocompatible silica-gelatin hybrid aerogels covalently labeled with fluorescein. J. Non-Cryst. Solids 2017, 473, 17–25. [Google Scholar] [CrossRef]
- Rezwan, K.; Chen, Q.Z.; Blaker, J.J.; Bocaccini, A.R. Biodegradable and bioactive porous polymer/inorganic composite scaffolds for bone tissue engineering. Biomaterials 2006, 27, 3413–3431. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Romer, F.; Connell, L.; Walter, C.; Saiz, E.; Yue, S.; Lee, P.D.; McPhail, D.S.; Hanna, J.V.; Jones, J.R. Highly flexible silica/chitosan hybrid scaffolds with oriented pores for tissue regeneration. J. Mater. Chem. B 2015, 3, 7560–7576. [Google Scholar] [CrossRef] [PubMed]
- Jerome, C.; Croisier, F. Chitosan-based biomaterials for tissue engineering. Eur. Polym. J. 2013, 49, 780–792. [Google Scholar] [CrossRef]
- Kechagias, S.; Moschogiannaki, F.; Stratakis, E.; Tzeranis, D.S.; Vosniakos, G.C. Porous collagen scaffold micro-fabrication: Feature-based process planning for computer numerically controlled laser systems. Int. J. Adv. Manuf. Technol. 2020, 111, 749–763. [Google Scholar] [CrossRef]
- Lucía, T.; Hern, A.C.; Rodríguez-lorenzo, L.M. Preparation of covalently bonded silica-alginate hybrid hydrogels by SCHIFF base and sol-gel reactions. Carbohydr. Polym. 2021, 267, 118186. [Google Scholar] [CrossRef]
- Wu, J.; Zheng, K.; Huang, X.; Liu, J.; Liu, H.; Boccaccini, A.R.; Wan, Y.; Guo, X.; Shao, Z. Thermally triggered injectable chitosan/silk fibroin/bioactive glass nanoparticle hydrogels for in-situ bone formation in rat calvarial bone defects. Acta Biomater. 2019, 91, 60–71. [Google Scholar] [CrossRef]
- Hollister, S.J. Porous scaffold design for tissue engineering. Nat. Mater. 2005, 4, 518–524. [Google Scholar] [CrossRef]
- Reyes-Peces, M.V.; Fernández-Montesinos, R.; del Mar Mesa-Díaz, M.; Vilches-Pérez, J.I.; Cárdenas-Leal, J.L.; de la Rosa-Fox, N.; Salido, M.; Piñero, M. Structure-Related Mechanical Properties and Bioactivity of Silica–Gelatin Hybrid Aerogels for Bone Regeneration. Gels 2023, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Mahony, O.; Tsigkou, O.; Ionescu, C.; Minelli, C.; Ling, L.; Hanly, R.; Smith, M.E.; Stevens, M.M.; Jones, J.R. Silica-gelatin hybrids with tailorable degradation and mechanical properties for tissue regeneration. Adv. Funct. Mater. 2010, 20, 3835–3845. [Google Scholar] [CrossRef]
- Dong, Y.; Liang, J.; Cui, Y.; Xu, S.; Zhao, N. Fabrication of novel bioactive hydroxyapatite-chitosan-silica hybrid scaffolds: Combined the sol-gel method with 3D plotting technique. Carbohydr. Polym. 2018, 197, 183–193. [Google Scholar] [CrossRef]
- Martínez-vázquez, F.J.; Cabañas, M.V.; Paris, J.L.; Lozano, D.; Vallet-regí, M. Fabrication of novel Si-doped hydroxyapatite/gelatine scaffolds by rapid prototyping for drug delivery and bone regeneration. Acta Biomater. 2015, 15, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, S.; Heinemann, C.; Wenisch, S.; Alt, V.; Worch, H.; Hanke, T. Calcium phosphate phases integrated in silica/collagen nanocomposite xerogels enhance the bioactivity and ultimately manipulate the osteoblast/osteoclast ratio in a human co-culture model. Acta Biomater. 2013, 9, 4878–4888. [Google Scholar] [CrossRef] [PubMed]
- Jayash, S.N.; Cooper, P.R.; Shelton, R.M.; Kuehne, S.A.; Poologasundarampillai, G. Novel chitosan-silica hybrid hydrogels for cell encapsulation and drug delivery. Int. J. Mol. Sci. 2021, 22, 12267. [Google Scholar] [CrossRef] [PubMed]
- Demilecamps, A.; Beauger, C.; Hildenbrand, C.; Rigacci, A.; Budtova, T. Cellulose-silica aerogels. Carbohydr. Polym. 2015, 122, 293–300. [Google Scholar] [CrossRef]
- García-González, C.A.; Budtova, T.; Dur, L.; Erkey, C.; Del Gaudio, P.; Gurikov, P.; Koebel, M.; Liebner, F.; Neagu, M.; Smirnova, I. An Opinion Paper on Aerogels for Biomedical and Environmental Applications. Molecules 2019, 24, 1815. [Google Scholar] [CrossRef]
- Bharadwaz, A.; Jayasuriya, A.C. Recent Trends in the Application of Widely Used Natural and Synthetic Polymer Nanocomposites in Bone Tissue Regeneration. Mater. Sci. Eng. C 2021, 110, 110698. [Google Scholar] [CrossRef]
- Roseti, L.; Parisi, V.; Petretta, M.; Cavallo, C.; Desando, G.; Bartolotti, I.; Grigolo, B. Scaffolds for Bone Tissue Engineering: State of the art and new perspectives. Mater. Sci. Eng. C 2017, 78, 1246–1262. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Y.; Zhou, Y. Application Progress of Modified Chitosan and Its Composite Biomaterials for Bone Tissue Engineering. Int. J. Mol. Sci. 2022, 23, 6574. [Google Scholar] [CrossRef] [PubMed]
- Celesti, C.; Iannazzo, D.; Espro, C.; Visco, A.; Legnani, L.; Veltri, L.; Visalli, G.; Di Pietro, A.; Bottino, P.; Chiacchio, M.A. Chitosan/POSS Hybrid Hydrogels for Bone Tissue Engineering. Materials 2022, 15, 8208. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhang, S.; Xie, C.; Wan, X.; Li, X.; Chen, K.; Zhao, G. Preparation of High Mechanical Strength Chitosan Nanofiber/NanoSiO2/PVA Composite Scaffolds for Bone Tissue Engineering Using Sol–Gel Method. Polymers 2022, 14, 2083. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Kim, J.; Kim, J.; Lee, J.; Sang, U.; Lee, E.; Kim, H. Enhanced mechanical properties and bone bioactivity of chitosan/silica membrane by functionalized-carbon nanotube incorporation. Compos. Sci. Technol. 2014, 96, 31–37. [Google Scholar] [CrossRef]
- Logithkumar, R.; Keshavnarayan, A.; Dhivya, S.; Chawla, A.; Saravanan, S.; Selvamurugan, N. A review of chitosan and its derivatives in bone tissue engineering. Carbohydr. Polym. 2016, 151, 172–188. [Google Scholar] [CrossRef]
- Rinki, K.; Dutta, P.K.; Hunt, A.J.; MacQuarrie, D.J.; Clark, J.H. Chitosan aerogels exhibiting high surface area for biomedical application: Preparation, characterization, and antibacterial study. Int. J. Polym. Mater. Polym. Biomater. 2011, 60, 988–999. [Google Scholar] [CrossRef]
- Perez_Moreno, A.; Reyes-Peces, M.V.; Vilches-Pérez, J.I.; Fernández-Montesinos, R.; Pinaglia-Tobaruela, G.; Salido, M.; de la Rosa-fox, N.; Piñero, M. Effect of Washing Treatment on the Textural Properties and Bioactivity of Silica/Chitosan/TCP Xerogels for Bone Regeneration. Int. J. Mol. Sci. 2021, 22, 8321. [Google Scholar] [CrossRef]
- Reyes-Peces, M.V.; Pérez-Moreno, A.; De-los-Santos, D.M.; del Mar Mesa-Díaz, M.; Pinaglia-Tobaruela, G.; Vilches-Pérez, J.I.; Fernández-Montesinos, R.; Salido, M.; de la Rosa-Fox, N.; Piñero, M. Chitosan-GPTMS-silica hybrid mesoporous aerogels for bone tissue engineering. Polymer 2020, 12, 2723. [Google Scholar] [CrossRef]
- Perez-Moreno, A.; Reyes-Peces, M.V.; de los Santos, D.M.; Pinaglia-Tobaruela, G.; de la Orden, E.; Vilches-Pérez, J.I.; Salido, M.; Piñero, M.; de la Rosa-Fox, N. Hydroxyl groups induce bioactivity in silica/chitosan aerogels designed for bone tissue engineering. In vitro model for the assessment of osteoblasts behavior. Polymers 2020, 12, 2802. [Google Scholar] [CrossRef]
- Trujillo, S.; Pérez-Román, E.; Kyritsis, A.; Gõmez Ribelles, J.L.; Pandis, C. Organic-inorganic bonding in chitosan-silica hybrid networks: Physical properties. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 1391–1400. [Google Scholar] [CrossRef]
- Palla-rubio, B.; Araújo-gomes, N.; Fernández-gutiérrez, M.; Rojo, L.; Suay, J.; Gurruchaga, M. Synthesis and characterization of silica-chitosan hybrid materials as antibacterial coatings for titanium implants. Carbohydr. Polym. 2019, 203, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Pipattanawarothai, A.; Suksai, C.; Srisook, K.; Trakulsujaritchok, T. Non-cytotoxic hybrid bioscaffolds of chitosan-silica: Sol-gel synthesis, characterization and proposed application. Carbohydr. Polym. 2017, 178, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Budnyak, T.M.; Pylypchuk, I.V.; Tertykh, V.A.; Yanovska, E.S.; Kolodynska, D. Synthesis and adsorption properties of chitosan-silica nanocomposite prepared by sol-gel method. Nanoscale Res.Lett. 2015, 10, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Alvarez Echazú, M.I.; Renou, S.J.; Alvarez, G.S.; Desimone, M.F.; Olmedo, D.G. Synthesis and Evaluation of a Chitosan–Silica-Based Bone Substitute for Tissue Engineering. Int. J. Mol. Sci. 2022, 23, 13379. [Google Scholar] [CrossRef] [PubMed]
- Toskas, G.; Cherif, C.; Hund, R.D.; Laourine, E.; Mahltig, B.; Fahmi, A.; Heinemann, C.; Hanke, T. Chitosan(PEO)/silica hybrid nanofibers as a potential biomaterial for bone regeneration. Carbohydr. Polym. 2013, 94, 713–722. [Google Scholar] [CrossRef]
- da Costa Neto, B.P.; da Mata, A.L.M.L.; Lopes, M.V.; Rossi-Bergmann, B.; Ré, M.I. Preparation and evaluation of chitosan-hydrophobic silica composite microspheres: Role of hydrophobic silica in modifying their properties. Powder Technol. 2014, 255, 109–119. [Google Scholar] [CrossRef]
- Connell, L.S.; Romer, F.; Suárez, M.; Valliant, E.M.; Zhang, Z.; Lee, P.D.; Smith, M.E.; Hanna, J.V.; Jones, J.R. Chemical characterisation and fabrication of chitosan-silica hybrid scaffolds with 3-glycidoxypropyl trimethoxysilane. J. Mater. Chem. B 2014, 2, 668–680. [Google Scholar] [CrossRef]
- Ayers, M.R.; Hunt, A.J. Synthesis and properties of chitosan-silica hybrid aerogels. J. Non-Cryst. Solids 2001, 285, 123–127. [Google Scholar] [CrossRef]
- Connell, L.S.; Gabrielli, L.; Mahony, O.; Russo, L.; Cipolla, L.; Jones, J.R. Functionalizing natural polymers with alkoxysilane coupling agents: Reacting 3-glycidoxypropyl trimethoxysilane with poly(γ-glutamic acid) and gelatin. Polym. Chem. 2017, 8, 1095–1103. [Google Scholar] [CrossRef]
- Balavigneswaran, C.K.; Venkatesan, R.; Karuppiah, P.S.; Kumar, G.; Paliwal, P.; Krishnamurthy, S.; Kadalmani, B.; Mahto, S.K.; Misra, N. Silica release from silane cross-linked gelatin based hybrid scaffold affects cell proliferation. ACS Appl. Bio Mater. 2020, 3, 197–207. [Google Scholar] [CrossRef]
- Fuentes, C.; Ruiz-Rico, M.; Fuentes, A.; Barat, J.M.; Ruiz, M.J. Comparative cytotoxic study of silica materials functionalised with essential oil components in HepG2 cells. Food Chem. Toxicol. 2021, 147, 111858. [Google Scholar] [CrossRef] [PubMed]
- Houmard, M.; Fu, Q.; Genet, M.; Saiz, E.; Tomsia, A.P. On the structural, mechanical, and biodegradation properties of HA/b -TCP robocast scaffolds. J. Biomed. Mater. Res. Part B 2013, 101, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Kuboyama, N. A biodegradable porous composite scaffold of PGA/β-TCP for bone tissue engineering. Bone 2010, 46, 386–395. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, Y.; Luo, J.; Wang, P.; Xiao, P.; Jiang, B. The Influence of Water Content on the Growth of the Hybrid-Silica Particles by Sol-Gel Method. Silicon 2021, 13, 3413–3421. [Google Scholar] [CrossRef]
- Buckley, A.M.; Greenblatt, M. A comparison of the microstructural properties of silica aerogels and xerogels. J. Non-Cryst. Solids 1992, 143, 1–13. [Google Scholar] [CrossRef]
- Canillas, M.; Pena, P.; De Aza, A.H.; Rodríguez, M.A. Calcium phosphates for biomedical applications. Bol. Soc. Esp. Ceram. Vidr. 2017, 56, 91–112. [Google Scholar] [CrossRef]
- Safronova, T.V.; Selezneva, I.I.; Tikhonova, S.A.; Kiselev, A.S.; Davydova, G.A.; Shatalova, T.B.; Larionov, D.S.; Rau, J.V. Biocompatibility of biphasic α,β-tricalcium phosphate ceramics in vitro. Bioact. Mater. 2020, 5, 423–427. [Google Scholar] [CrossRef]
- Gregg, S.J.; Sing, K.S.W. Adsorption, Surface Area and Porosity, 2nd ed.; Gregg, S.J., Ed.; Academic Press: London, UK, 1982. [Google Scholar]
- Sing, K. Reporting physisorption data for gas/solid systems with Special Reference to the Determination of Surface Area and Porosity. Pure Appl. Chem. 1982, 54, 2201–2218. [Google Scholar] [CrossRef]
- Galarneau, A.; Mehlhorn, D.; Guenneau, F.; Coasne, B.; Villemot, F.; Minoux, D.; Aquino, C.; Dath, J. Specific Surface Area Determination for Microporous/Mesoporous Materials: The Case of Mesoporous FAU-Y Zeolites. Langmuir 2018, 34, 14134–14142. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Williams, R.T. Physisorption hysteresis loops and the characterization of nanoporous materials. Adsorpt. Sci. Technol. 2004, 22, 773–782. [Google Scholar] [CrossRef]
- Smitha, S.; Shajesh, P.; Mukundan, P.; Warrier, K.G.K. Sol-gel synthesis of biocompatible silica-chitosan hybrids and hydrophobic coatings. J. Mater. Res. 2008, 23, 2053–2060. [Google Scholar] [CrossRef]
- Jinlong, N.; Zhenxi, Z.; Dazong, J. Investigation of Phase Evolution During the Thermochemical Synthesis of Tricalcium Phosphate. J. Mater. Synth. Process. 2001, 9, 235–240. [Google Scholar] [CrossRef]
- Venkateswara Rao, A.; Kalesh, R.R. Comparative studies of the physical and hydrophobic properties of TEOS based silica aerogels using different co-precursors. Sci. Technol. Adv. Mater. 2003, 4, 509–515. [Google Scholar] [CrossRef]
- Biggs, M.J.P.; Dalby, M.J. Focal adhesions in osteoneogenesis. Proc. Inst. Mech. Eng. H 2010, 224, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Bačáková, L.; Filová, E.; Rypáček, F.; Švorčík, V.; Starý, V. Cell Adhesion on Artificial Materials for Tissue Engineering. Physiol. Res. 2004, 53, S35–S45. [Google Scholar] [CrossRef] [PubMed]
- Terriza, A.; Vilches-Pérez, J.I.; González-Caballero, J.L.; de la Orden, E.; Yubero, F.; Barranco, A.; Gonzalez-Elipe, A.R.; Vilches, J.; Salido, M. Osteoblasts Interaction with PLGA Membranes Functionalized with Titanium Film Nanolayer by PECVD. Materials 2014, 7, 1687–1708. [Google Scholar] [CrossRef] [PubMed]
- Coyer, S.R.; Singh, A.; Dumbauld, D.W.; Calderwood, D.A.; Craig, S.W.; Delamarche, E.; García, A.J. Nanopatterning reveals an ECM area threshold for focal adhesion assembly and force transmission that is regulated by integrin activation and cytoskeleton tension. J. Cell Sci. 2012, 125, 5110–5123. [Google Scholar] [CrossRef]
- Bays, J.L.; DeMali, K.A. Vinculin in cell–cell and cell–matrix adhesions. Cell. Mol. Life Sci. 2017, 74, 2999–3009. [Google Scholar] [CrossRef]
- Natale, C.F.; Ventre, M.; Netti, P.A. Tuning the material-cytoskeleton crosstalk via nanocon fi nement of focal adhesions. Biomaterials 2014, 35, 2743–2751. [Google Scholar] [CrossRef]
- Salido, M.; Vilches, J.I.; Gutiérrez, J.L.; Vilches, J. Actin cytoskeletal organization in human osteoblasts grown on different dental titanium implant surfaces. Histol. Histopathol. 2007, 22, 1355–1364. [Google Scholar] [CrossRef]
- Salido, M.; Vilches-perez, J.I.; Gonzalez, J.L.; Vilches, J. Mitochondrial bioenergetics and distribution in living human osteoblasts grown on implant surfaces. Histol. Histopathol. 2009, 24, 1275–1286. [Google Scholar] [PubMed]
- Lamers, E.; van Horssen, R.; te Riet, J.; van Delft, F.C.M.J.M.; Luttge, R.; Walboomers, X.F.; Jansen, J.A. The influence of nanoscale topographical cues on initial osteoblast morphology and migration. Eur. Cells Mater. 2010, 20, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Jonathan, M.; Biggs, P.; Richards, R.G.; Dalby, M.J. Nanotopographical modification: A regulator of cellular function through focal adhesions. Nanomedicine 2010, 6, 619–633. [Google Scholar] [CrossRef]
- Vallet-Regí, M.; Feito, M.J.; Arcos, D.; Oñaderra, M.; Matesanz, M.C.; Martínez-Vázquez, F.J.; Sánchez-Salcedo, S.; Portolés, M.T.; Linares, J. Response of osteoblasts and preosteoblasts to calcium deficient and Si substituted hydroxyapatites treated at different temperatures. Colloids Surf. B Biointerfaces 2015, 133, 304–313. [Google Scholar] [CrossRef]
- Owens, G.J.; Singh, R.K.; Foroutan, F.; Alqaysi, M.; Han, C.M.; Mahapatra, C.; Kim, H.W.; Knowles, J.C. Sol-gel based materials for biomedical applications. Prog. Mater. Sci. 2016, 77, 1–79. [Google Scholar] [CrossRef]
- Serra, I.R.; Fradique, R.; Vallejo, M.C.S.; Correia, T.R.; Miguel, S.P.; Correia, I.J. Production and characterization of chitosan/gelatin/β-TCP scaffolds for improved bone tissue regeneration. Mater. Sci. Eng. C 2015, 55, 592–604. [Google Scholar] [CrossRef]
- Arcos, D.; Vallet-regí, M. Acta Biomaterialia Sol-gel silica-based biomaterials and bone tissue regeneration. Acta Biomater. 2010, 6, 2874–2888. [Google Scholar] [CrossRef]
- Bittig, A.T.; Matschegewski, C.; Nebe, J.B.; Stählke, S.; Uhrmacher, A.M. Membrane related dynamics and the formation of actin in cells growing on micro-topographies: A spatial computational model. BNC Syst. Biol. 2014, 8, 106–125. [Google Scholar] [CrossRef]
- Mullen, C.A.; Vaughan, T.J.; Voisin, M.C.; Brennan, M.A.; Layrolle, P.; McNamara, L.M. Cell morphology and focal adhesion location alters internal cell stress. J. R. Soc. Interface 2014, 11, 1–12. [Google Scholar] [CrossRef]
- Tang, Z.; Li, X.; Tan, Y.; Fan, H.; Zhang, X. The material and biological characteristics of osteoinductive calcium phosphate ceramics. Regen. Biomater. 2018, 5, 43–59. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ρ * (gcm−3) | Volume Shrinkage * (%) | SBET ** (m2g−1) | Vp (cm3g−1) | Pore Size (nm) |
|---|---|---|---|---|---|
| SiO2A | 0.19 ± 0.01 | 33.0 ± 2.3 | 978.2 | 4.1 | 16.9 |
| SCS8A | 0.18 ± 0.03 | 30.2 ± 3.1 | 857.7 | 3.9 | 17.3 |
| SiO2X | 0.61 ± 0.05 | 75.7 ± 2.6 | 807.5 | 1.0 | 4.7 |
| SCS8X | 0.49 ± 0.10 | 72.3 ± 4.2 | 821.1 | 1.1 | 5.0 |
| SCS8T10X | 0.54 ± 0.03 | 67.4 ± 2.4 | 733.6 | 1.5 | 7.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Moreno, A.; Piñero, M.; Fernández-Montesinos, R.; Pinaglia-Tobaruela, G.; Reyes-Peces, M.V.; Mesa-Díaz, M.d.M.; Vilches-Pérez, J.I.; Esquivias, L.; de la Rosa-Fox, N.; Salido, M. Chitosan-Silica Hybrid Biomaterials for Bone Tissue Engineering: A Comparative Study of Xerogels and Aerogels. Gels 2023, 9, 383. https://doi.org/10.3390/gels9050383
Pérez-Moreno A, Piñero M, Fernández-Montesinos R, Pinaglia-Tobaruela G, Reyes-Peces MV, Mesa-Díaz MdM, Vilches-Pérez JI, Esquivias L, de la Rosa-Fox N, Salido M. Chitosan-Silica Hybrid Biomaterials for Bone Tissue Engineering: A Comparative Study of Xerogels and Aerogels. Gels. 2023; 9(5):383. https://doi.org/10.3390/gels9050383
Chicago/Turabian StylePérez-Moreno, Antonio, Manuel Piñero, Rafael Fernández-Montesinos, Gonzalo Pinaglia-Tobaruela, María V. Reyes-Peces, María del Mar Mesa-Díaz, José Ignacio Vilches-Pérez, Luis Esquivias, Nicolás de la Rosa-Fox, and Mercedes Salido. 2023. "Chitosan-Silica Hybrid Biomaterials for Bone Tissue Engineering: A Comparative Study of Xerogels and Aerogels" Gels 9, no. 5: 383. https://doi.org/10.3390/gels9050383