
Metagenomic/Metaproteomic Investigation of the Microbiota in Dongbei Suaicai, a Traditional Fermented Chinese Cabbage
 , ,
, ,
Abstract
:Highlights
- Important metabolic pathways related to carbohydrate and amino acid metabolisms were enriched.
- Pediococcus was first found to be one of the most popular genera of late fermentation.
- The multi-omics of metagenomics and metaproteomics was used to research the microbial ecosystem of Dongbei Suaicai.
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Collection
2.2. Metagenomic Analysis
2.2.1. DNA Extraction, Library Construction, and Sequencing
2.2.2. Information-Analysis Process
2.3. Protein Preparation and Metaproteomics
2.3.1. Protein Extraction and Peptide Digestion
2.3.2. Mass Spectrometric Methods
2.3.3. MS Data Analysis
2.4. Bioinformatics Analysis
3. Results
3.1. Overview of Metagenomic Data
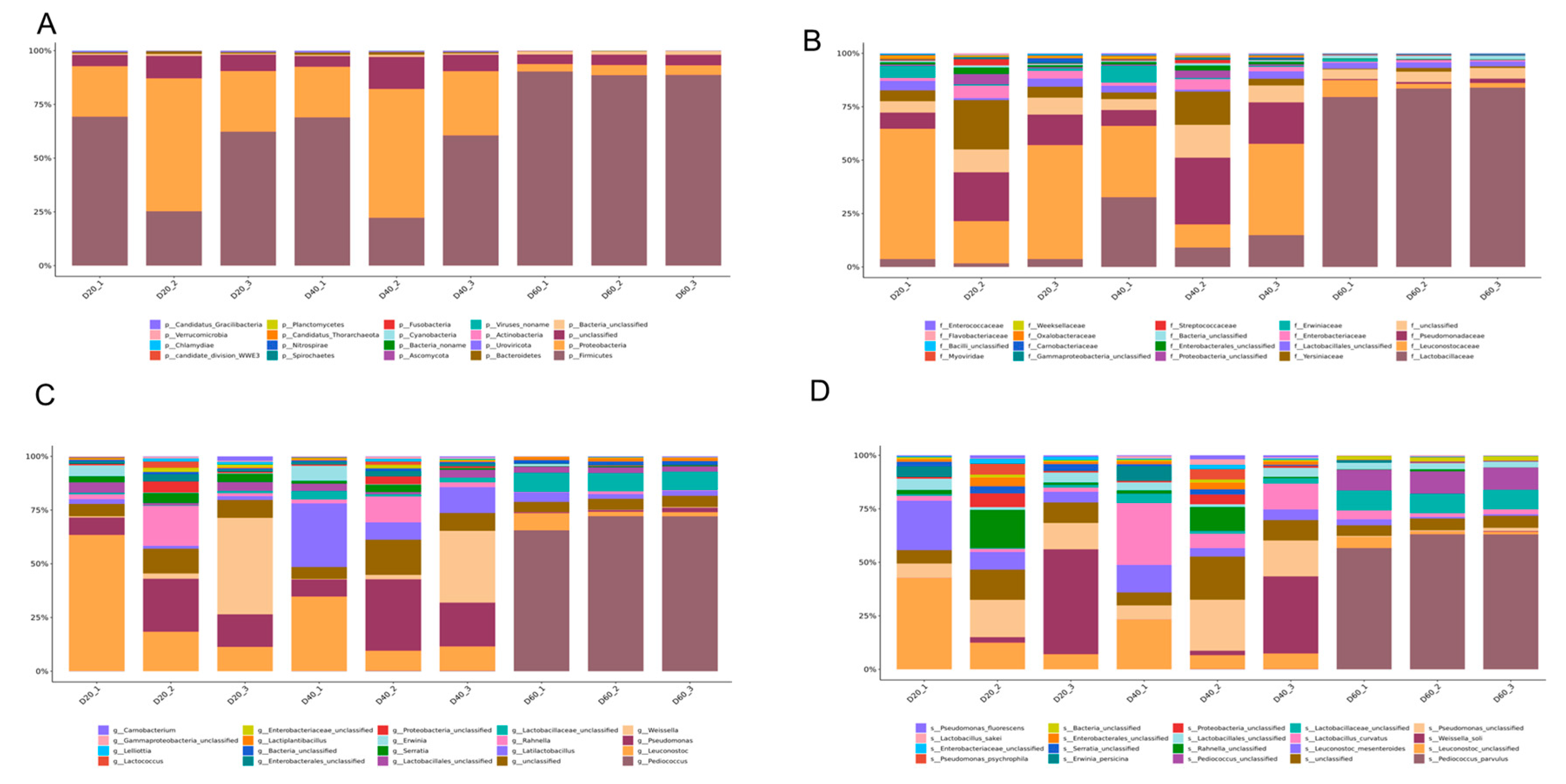
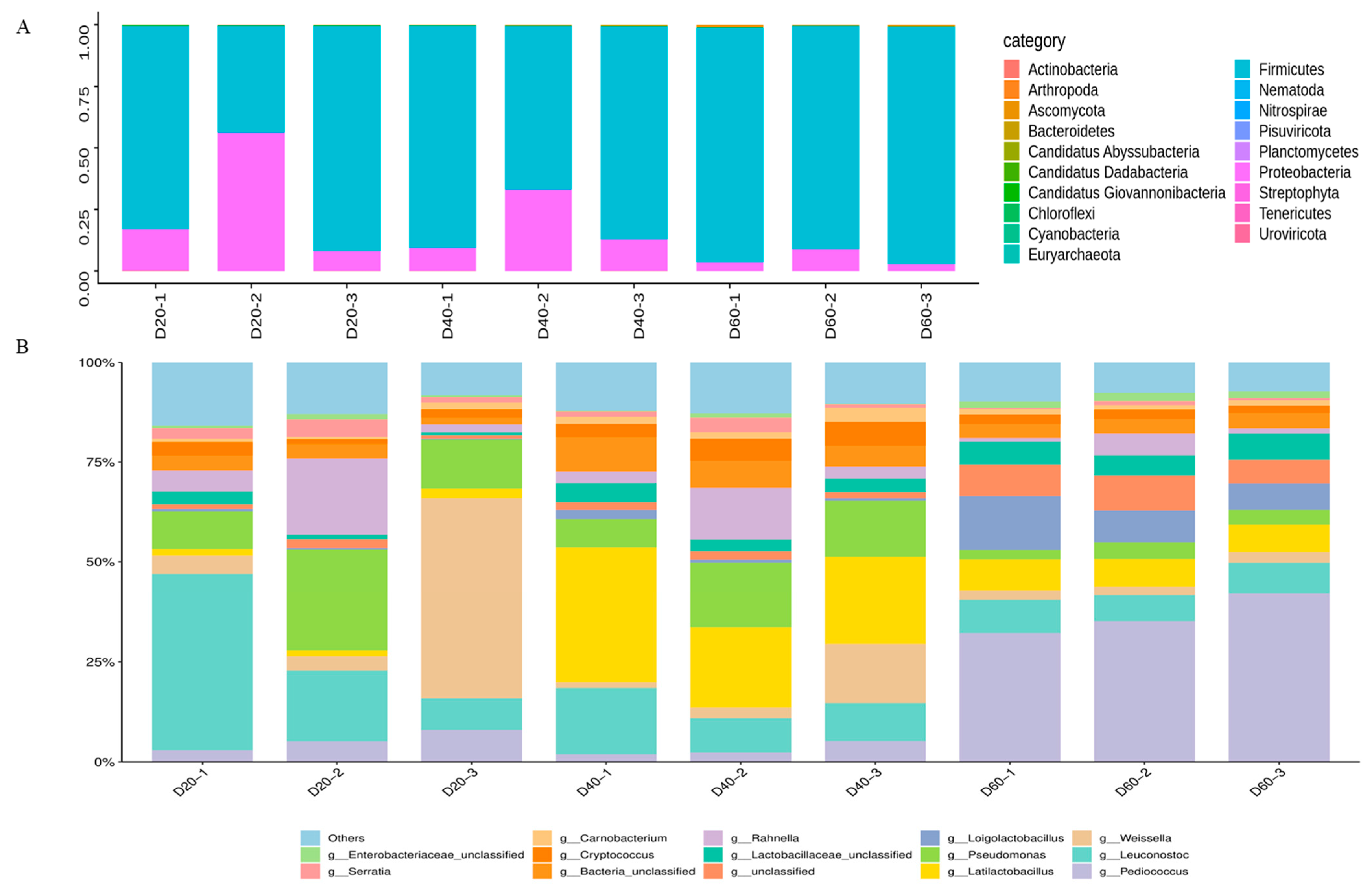
3.2. Taxonomic Composition of Predicted Genes
3.3. Function Classification of Identified Genes
3.4. Taxonomic Composition of Identified Proteins
3.5. Function Classification of Proteins
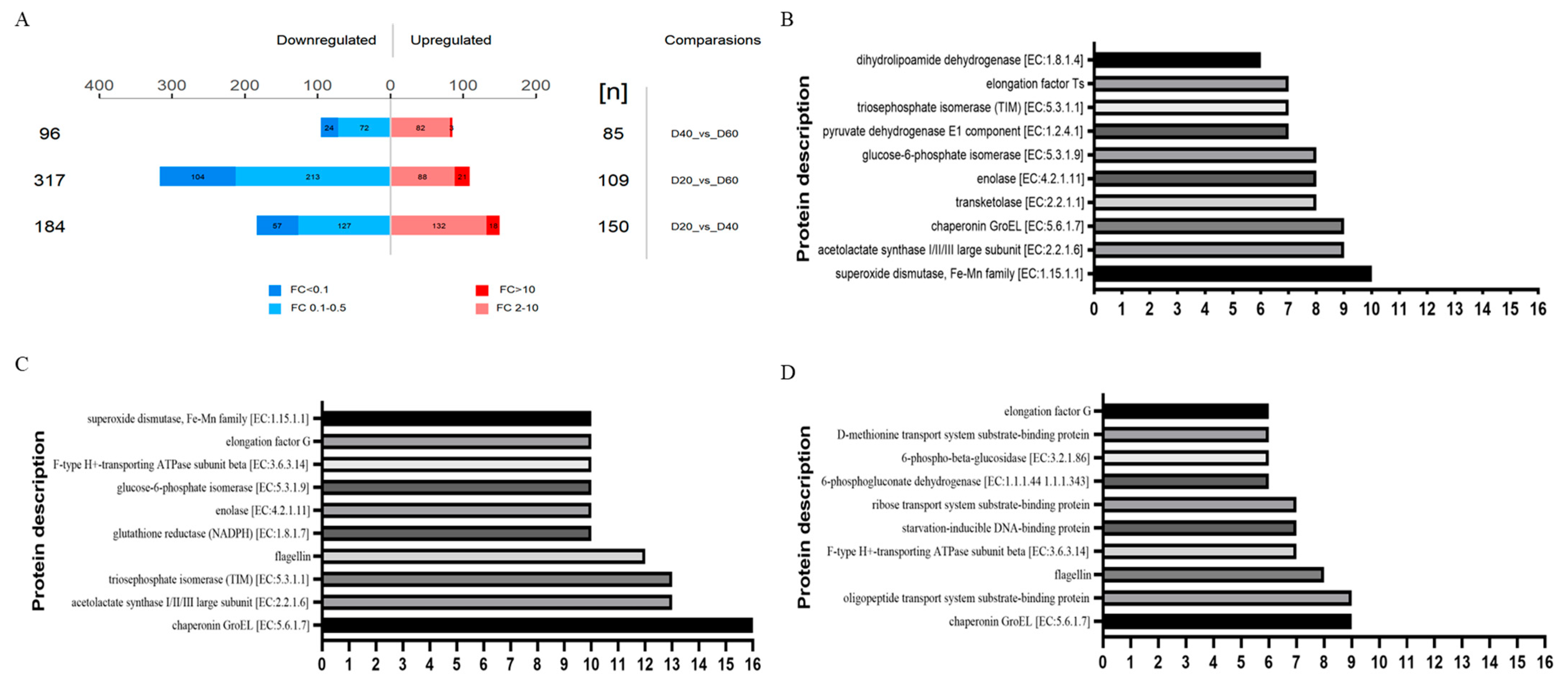
3.6. Overview and Analysis of Differential Proteins
4. Discussion
4.1. Microbial Community Composition
4.2. Biological Function Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ye, H.Q.; Lang, X.S.; Ji, Y.Y.; Li, S.N.; Xin, N.C.; Meng, X.R.; Zhang, T.H.; Shen, X.; Zhao, C.H. The interaction between Lactobacillus plantarum SC-5 and its biogenic amine formation with different salt concentrations in Chinese Dongbei Suancai. Food Res. Int. 2021, 150, 110813. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, A.; Raju, P.S.; Bawa, A.S. A Two-Step Controlled Lactic Fermentation of Cabbage for Improved Chemical and Microbiological Qualities. J. Food Qual. 2012, 35, 13–20. [Google Scholar] [CrossRef]
- Beganovic, J.; Kos, B.; Pavunc, A.L.; Uroic, K.; Jokic, M.; Suskovic, J. Traditionally produced sauerkraut as source of autochthonous functional starter cultures. Microbiol. Res. 2014, 169, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Huang, T.; Huang, C.; Hardie, J.; Peng, Z.; Xie, M.; Xiong, T. The microbial communities and flavour compounds of Jiangxi yancai, Sichuan paocai and Dongbei suancai: Three major types of traditional Chinese fermented vegetables. Lwt-Food Sci. Technol. 2020, 121, 108865. [Google Scholar] [CrossRef]
- Yang, X.; Hu, W.; Xiu, Z.; Jiang, A.; Yang, X.; Saren, G.; Ji, Y.; Guan, Y.; Feng, K. Effect of salt concentration on microbial communities, physicochemical properties and metabolite profile during spontaneous fermentation of Chinese northeast sauerkraut. J. Appl. Microbiol. 2020, 129, 1458–1471. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.X.; An, F.Y.; Wu, J.R.; Liu, Y.M.; Shi, H.S.; Wu, R.N. Meta-omics reveal microbial assortments and key enzymes in bean sauce mash, a traditional fermented soybean product. J. Sci. Food Agric. 2019, 99, 6522–6534. [Google Scholar] [CrossRef]
- Srinivas, M.; O’Sullivan, O.; Cotter, P.D.; van Sinderen, D.; Kenny, J.G. The Application of Metagenomics to Study Microbial Communities and Develop Desirable Traits in Fermented Foods. Foods 2022, 11, 3297. [Google Scholar] [CrossRef]
- Okeke, E.S.; Ita, R.E.; Egong, E.J.; Udofia, L.E.; Mgbechidinma, C.L.; Akan, O.D. Metaproteomics insights into fermented fish and vegetable products and associated microbes. Food Chem. Mol. Sci. 2021, 3, 100045. [Google Scholar] [CrossRef] [PubMed]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis. Evol. Bioinform. 2016, 12, 5–16. [Google Scholar] [CrossRef]
- Athanasopoulou, K.; Boti, M.A.; Adamopoulos, P.G.; Skourou, P.C.; Scorilas, A. Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life 2022, 12, 30. [Google Scholar] [CrossRef]
- Zhou, Z.; Tran, P.Q.; Breister, A.M.; Liu, Y.; Kieft, K.; Cowley, E.S.; Karaoz, U.; Anantharaman, K. METABOLIC: High-throughput profiling of microbial genomes for functional traits, metabolism, biogeochemistry, and community-scale functional networks. Microbiome 2022, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Jansson, J.K.; Hofmockel, K.S. The soil microbiome-from metagenomics to metaphenomics. Curr. Opin. Microbiol. 2018, 43, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Tan, Y.; Hong, H.; Li, D.; Zhang, L.; Luo, Y. Exploration of the roles of spoilage bacteria in degrading grass carp proteins during chilled storage: A combined metagenomic and metabolomic approach. Food Res. Int. 2022, 152, 110926. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Schmid, E.; de Castro, J.V.; Cardinale, M.; Eberl, L.; Grube, M.; Berg, G.; Riedel, K. Structure and function of the symbiosis partners of the lung lichen (Lobaria pulmonaria L. Hoffm.) analyzed by metaproteomics. Proteomics 2011, 11, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Riffle, M.; May, D.H.; Timmins-Schiffman, E.; Mikan, M.P.; Jaschob, D.; Noble, W.S.; Nunn, B.L. MetaGOmics: A Web-Based Tool for Peptide-Centric Functional and Taxonomic Analysis of Metaproteomics Data. Proteomes 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Bahule, C.E.; da Silva Martins, L.H.; Mussengue Chauque, B.J.; Lopes, A.S. Metaproteomics as a tool to optimize the maize fermentation process. Trends Food Sci. Technol. 2022, 129, 258–265. [Google Scholar] [CrossRef]
- Xia, Y.; Zhu, M.; Du, Y.K.; Wu, Z.Y.; Gomi, K.; Zhang, W.X. Metaproteomics reveals protein composition of multiple saccharifying enzymes in nongxiangxing daqu and jiangxiangxing daqu under different thermophilic temperatures. Int. J. Food Sci. Technol. 2022, 57, 5102–5113. [Google Scholar] [CrossRef]
- Muth, T.; Renard, B.Y.; Martens, L. Metaproteomic data analysis at a glance: Advances in computational microbial community proteomics. Expert Rev. Proteom. 2016, 13, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Okuda, S. Current progress and critical challenges to overcome in the bioinformatics of mass spectrometry-based metaproteomics. Comput. Struct. Biotechnol. J. 2023, 21, 1140–1150. [Google Scholar] [CrossRef]
- Wu, F.; Ding, X.; Zhang, Y.; Gu, J.-D.; Liu, X.; Guo, Q.; Li, J.; Feng, H. Metagenomic and metaproteomic insights into the microbiome and the key geobiochemical potentials on the sandstone of rock-hewn Beishiku Temple in Northwest China. Sci. Total Environ. 2023, 893, 164616. [Google Scholar] [CrossRef]
- Xie, M.; Wu, J.; An, F.; Yue, X.; Tao, D.; Wu, R.; Lee, Y. An integrated metagenomic/metaproteomic investigation of microbiota in dajiang-meju, a traditional fermented soybean product in Northeast China. Food Res. Int. 2019, 115, 414–424. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, C.; Zhang, J.; Xin, X.; Liao, X. Metagenomics reveals the formation mechanism of flavor metabolites during the spontaneous fermentation of potherb mustard (Brassica juncea var. multiceps). Food Res. Int. 2021, 148, 110622. [Google Scholar] [CrossRef]
- He, J.; Li, F.; Wang, Y.; Wu, H.; Yang, H. Fermentation characteristics and bacterial dynamics during Chinese sauerkraut fermentation by Lactobacillus curvatus LC-20 under varied salt concentrations reveal its potential in low-salt suan cai production. J. Biosci. Bioeng. 2021, 132, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Li, J.Y.; Wei, B.L.; Huang, T.; Xiao, Y.S.; Peng, Z.; Xie, M.Y.; Xiong, T. Bacterial community and composition in Jiang-shui and Suan-cai revealed by high-throughput sequencing of 16S rRNA. Int. J. Food Microbiol. 2019, 306, 108271. [Google Scholar] [CrossRef]
- Li, Q.; Kang, J.; Ma, Z.; Li, X.; Liu, L.; Hu, X. Microbial succession and metabolite changes during traditional serofluid dish fermentation. Lwt-Food Sci. Technol. 2017, 84, 771–779. [Google Scholar] [CrossRef]
- Yang, X.; Hu, W.; Xiu, Z.; Jiang, A.; Yang, X.; Saren, G.; Ji, Y.; Guan, Y.; Feng, K. Microbial Community Dynamics and Metabolome Changes During Spontaneous Fermentation of Northeast Sauerkraut From Different Households. Front. Microbiol. 2020, 11, 1878. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xie, S.; Sun, B.; Zhang, Y.; Liu, K.; Liu, L. Effect of KCl replacement of NaCl on fermentation kinetics, organic acids and sensory quality of sauerkraut from Northeast China. J. Food Process. Preserv. 2022, 46, e16622. [Google Scholar] [CrossRef]
- Suissa, R.; Oved, R.; Maan, H.; Hadad, U.; Gilhar, O.; Meijler, M.M.; Koren, O.; Kolodkin-Gal, I. Context-dependent differences in the functional responses of Lactobacillaceae strains to fermentable sugars. Front. Microbiol. 2022, 13, 949932. [Google Scholar] [CrossRef]
- Kim, E.; Yang, S.-M.; Kim, I.-S.; Lee, S.-Y.; Kim, H.-Y. Identification of Leuconostoc species based on novel marker genes identified using real-time PCR via computational pangenome analysis. Front. Microbiol. 2022, 13, 1014872. [Google Scholar] [CrossRef]
- Yang, X.; Hu, W.; Xiu, Z.; Jiang, A.; Yang, X.; Sarengaowa; Ji, Y.; Guan, Y.; Feng, K. Microbial dynamics and volatilome profiles during the fermentation of Chinese northeast sauerkraut by Leuconostoc mesenteroides ORC 2 and Lactobacillus plantarum HBUAS 51041 under different salt concentrations. Food Res. Int. 2020, 130, 108926. [Google Scholar] [CrossRef]
- Zabat, M.A.; Sano, W.H.; Wurster, J.I.; Cabral, D.J.; Belenky, P. Microbial Community Analysis of Sauerkraut Fermentation Reveals a Stable and Rapidly Established Community. Foods 2018, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Alexzman, Z.A.; Ibrahim, M.L.; Annuar, N.H.R. A short review on catalytic hydrogenation of fructose into mannitol. J. Chem. Technol. Biotechnol. 2023, 98, 2618–2626. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Wu, L.; Zhang, L.; Wang, S. Characterization of microbiota of naturally fermented sauerkraut by high-throughput sequencing. Food Sci. Biotechnol. 2023, 32, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Tlais, A.Z.A.; Lemos, W.J.F., Jr.; Filannino, P.; Campanaro, S.; Gobbetti, M.; Di Cagno, R. How Microbiome Composition Correlates with Biochemical Changes during Sauerkraut Fermentation: A Focus on Neglected Bacterial Players and Functionalities. Microbiol. Spectr. 2022, 10, e00168-22. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; She, X.; Chen, X.; Qian, Y.; Tao, Y.; Li, Y.; Guo, S.; Xiang, W.; Liu, G.; Rao, Y. Microbiota Succession and Chemical Composition Involved in the Radish Fermentation Process in Different Containers. Front. Microbiol. 2020, 11, 502490. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Shen, Q.; Liu, Z.; Fu, P.; Zhou, W. A newly isolated strain Pediococcus parvulus from Xuanwei ham, a traditional Chinese fermented meat product. Int. J. Food Sci. Technol. 2008, 43, 1387–1394. [Google Scholar] [CrossRef]
- Maharachchikumbura, S.S.N.; Chen, Y.P.; Ariyawansa, H.A.; Hyde, K.D.; Haelewaters, D.; Perera, R.H.; Samarakoon, M.C.; Wanasinghe, D.N.; Bustamante, D.E.; Liu, J.K.; et al. Integrative approaches for species delimitation in Ascomycota. Fungal Divers. 2021, 109, 155–179. [Google Scholar] [CrossRef]
- Barrangou, R.; Yoon, S.S.; Breidt, F.; Fleming, H.P.; Klaenhammer, T.R. Identification and characterization of Leuconostoc fallax strains isolated from an industrial sauerkraut fermentation. Appl. Environ. Microbiol. 2002, 68, 2877–2884. [Google Scholar] [CrossRef] [PubMed]
- Maske, B.L.; Pereira, G.V.d.M.; Neto, D.P.d.C.; Lindner, J.d.D.; Letti, L.A.J.; Pagnoncelli, M.G.; Soccol, C.R. Presence and persistence of Pseudomonas sp. during Caspian Sea-style spontaneous milk fermentation highlights the importance of safety and regulatory concerns for traditional and ethnic foods. Food Sci. Technol. 2021, 41, 273–283. [Google Scholar] [CrossRef]
- Rawls, K.D.M. Organizational Readiness, Resistance and Race: Examining Faculty Attitudes toward Faculty Diversification in Higher Education. Ph.D. Thesis, Claremont Graduate University, Claremont, CA, USA, 2020. [Google Scholar]
- Wang, D.; Cui, F.; Ren, L.; Tan, X.; Lv, X.; Li, Q.; Li, J.; Li, T. Complete Genome Analysis Reveals the Quorum Sensing-Related Spoilage Potential of Pseudomonas fluorescens PF08, a Specific Spoilage Organism of Turbot (Scophthalmus maximus). Front. Microbiol. 2022, 13, 856802. [Google Scholar] [CrossRef]
- Parlapani, F.F.; Anagnostopoulos, D.A.; Karamani, E.; Mallouchos, A.; Haroutounian, S.A.; Boziaris, I.S. Growth and Volatile Organic Compound Production of Pseudomonas Fish Spoiler Strains on Fish Juice Agar Model Substrate at Different Temperatures. Microorganisms 2023, 11, 189. [Google Scholar] [CrossRef]
- Cleto, S.; Matos, S.; Kluskens, L.; Vieira, M.J. Characterization of Contaminants from a Sanitized Milk Processing Plant. PLoS ONE 2012, 7, e40189. [Google Scholar] [CrossRef]
- Du, B.; Lu, M.; Liu, H.; Wu, H.; Zheng, N.; Zhang, Y.; Zhao, S.; Zhao, Y.; Gao, T.; Wang, J. Pseudomonas isolates from raw milk with high level proteolytic activity display reduced carbon substrate utilization and higher levels of antibiotic resistance. Lwt-Food Sci. Technol. 2023, 181, 114766. [Google Scholar] [CrossRef]
- Fusco, V.; Quero, G.M.; Cho, G.-S.; Kabisch, J.; Meske, D.; Neve, H.; Bockelmann, W.; Franz, C.M.A.P. The genus Weissella: Taxonomy, ecology and biotechnological potential. Front. Microbiol. 2015, 6, 130276. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, Y.; Chen, L.; Zheng, J.; Lv, X.; Li, D.; Fang, Z.; Shen, C.; Mallawaarachchi, V.; Lin, Y.; et al. Quantitative metaproteomics reveals composition and metabolism characteristics of microbial communities in Chinese liquor fermentation starters. Front. Microbiol. 2023, 13, 1098268. [Google Scholar] [CrossRef]
- Lohmann, P.; Schäpe, S.S.; Haange, S.B.; Oliphant, K.; Allen-Vercoe, E.; Jehmlich, N.; Von Bergen, M. Function is what counts: How microbial community complexity affects species, proteome and pathway coverage in metaproteomics. Expert Rev. Proteom. 2020, 17, 163–173. [Google Scholar] [CrossRef]
- Escalas, A.; Hale, L.; Voordeckers, J.W.; Yang, Y.; Firestone, M.K.; Alvarez-Cohen, L.; Zhou, J. Microbial functional diversity: From concepts to applications. Ecol. Evol. 2019, 9, 12000–12016. [Google Scholar] [CrossRef] [PubMed]
- Bezerra do Ó, I.J.; Rego, T.G.; Jose, M.V.; de Farias, S.T. The structural proteome for the primordial glycolysis/gluconeogenesis. Genet. Mol. Res. 2020, 19, gmr18699. [Google Scholar] [CrossRef]
- Wieczorek, M.N.; Drabinska, N. Flavour Generation during Lactic Acid Fermentation of Brassica Vegetables-Literature Review. Appl. Sci. 2022, 12, 5598. [Google Scholar] [CrossRef]
- Moon, S.H.; Kim, C.R.; Chang, H.C. Heterofermentative lactic acid bacteria as a starter culture to control kimchi fermentation. Lwt-Food Sci. Technol. 2018, 88, 181–188. [Google Scholar] [CrossRef]
- Blajman, J.E.; Vinderola, G.; Paez, R.B.; Signorini, M.L. The role of homofermentative and heterofermentative lactic acid bacteria for alfalfa silage: A meta-analysis. J. Agric. Sci. 2020, 158, 107–118. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Li, C.; Zhao, Y.; Xiang, H.; Li, L.; Yang, X.; Chen, S.; Sun, L.; Qi, B. Genome-Resolved Metaproteomic Analysis of Microbiota and Metabolic Pathways Involved in Taste Formation during Chinese Traditional Fish Sauce (Yu-lu) Fermentation. Front. Nutr. 2022, 9, 851895. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q. Practical implications of lactate and pyruvate metabolism by lactic acid bacteria in food and beverage fermentations. Int. J. Food Microbiol. 2003, 83, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Dynesen, J.; Smits, H.P.; Olsson, L.; Nielsen, J. Carbon catabolite repression of invertase during batch cultivations of Saccharomyces cerevisiae: The role of glucose, fructose, and mannose. Appl. Microbiol. Biotechnol. 1998, 50, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Xiong, T.; Peng, Z.; Xiao, Y.-S.; Liu, Z.-G.; Hu, M.; Xie, M.-Y. Genomic analysis revealed adaptive mechanism to plant-related fermentation of Lactobacillus plantarum NCU116 and Lactobacillus spp. Genomics 2020, 112, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, X.; Li, F.; Shi, H.; He, M.; Ge, J.; Ling, H.; Cheng, K. Analysis of Microbial Diversity and Metabolites in Sauerkraut Products with and without Microorganism Addition. Foods 2023, 12, 1164. [Google Scholar] [CrossRef] [PubMed]
- Smit, G.; Smit, B.A.; Engels, W.J.M. Flavour formation by lactic acid bacteria and biochemical flavour profiling of cheese products. Fems Microbiol. Rev. 2005, 29, 591–610. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Z.; Hu, W.Z.; Xiu, Z.L.; Jiang, A.L.; Yang, X.Y.; Sarengaowa; Ji, Y.R.; Guan, Y.G.; Feng, K. Comparison of northeast sauerkraut fermentation between single lactic acid bacteria strains and traditional fermentation. Food Res. Int. 2020, 137, 109553. [Google Scholar] [CrossRef]
- Cartea, M.E.; Francisco, M.; Soengas, P.; Velasco, P. Phenolic Compounds in Brassica Vegetables. Molecules 2011, 16, 251–280. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eigenvalue | Data |
|---|---|
| Sequencing | |
| Raw reads(num) | 699,857,802 |
| Raw bases(bp) | 104,978,670,300 |
| Q20(%) | 96.77 |
| Q30(%) | 91.24 |
| Filter reads(num) | 683,407,618 |
| Filter bases(bp) | 101,969,988,159 |
| Q20(%) | 97.09 |
| Q30(%) | 91.64 |
| Valid% | 97.18 |
| Assembly | |
| Contigs (>=1000 bp) | 77,054 |
| Contigs (>=5000 bp) | 2302 |
| Contigs (>=10,000 bp) | 651 |
| Contigs (>=25,000 bp) | 158 |
| Contigs (>=50,000 bp) | 35 |
| Total length (>=25,000 bp) | 6,798,282 |
| Total length (>=50,000 bp) | 2,736,588 |
| Contigs | 241,723 |
| Largest contig | 163,143 |
| Total length | 262,961,360 |
| GC (%) | 52.75 |
| N50 | 1143 |
| N75 | 736 |
| L50 | 59,782 |
| L75 | 132,629 |
| Annotation | |
| ORFs NO. | 1,006,177 |
| Integrity start | 165,987 (16.50%) |
| Integrity end | 307,836 (30.59%) |
| Integrity all | 445,458 (44.27%) |
| Integrity none | 86,896 (8.64%) |
| Total Length (bp) | 649.6 |
| Average Length (bp) | 645.61 |
| GC percent | 54.96% |
| DEP | DEPperid | KO | KEGG PATHWAY | Main Species |
|---|---|---|---|---|
| oppA | D40vsD60 | K15580 | map01501, map02010, map02024 | Pediococcus |
| rbsB | D40vsD60 | K10439 | map02010, map02030 | Pseudomonas |
| SOD2 | D20vsD40 D20vsD60 | K04564 | map04013, map04068, map04146, map04211, map04212, map04213, map05016 | |
| groEL | D20vsD40 D40vsD60 D20vsD60 | K04077 | map03018, map04212, map04940, map05134, map05152 | Xanthomonas |
| dps | D40vsD60 | Serratia | ||
| fliC | D40vsD60 D20vsD60 | K02406 | map02020, map02040, map04621, map04626, map05132, map05134 | Pseudomonas |
| tsf | D20vsD40 | K02357 | map03018 | Pseudomonas |
| fusA | D40vsD60 D20vsD60 | K02112 | Pseudomonas | |
| atpD | D40vsD60 D20vsD60 | K02112 | map00190, map00195, map01100 | Pseudomonas |
| metQ | D40vsD60 | K02073 | map02010 | Weissella Rahnella |
| pgi | D20vsD40 D20vsD60 | K01810 | map00010, map00030, map00500, map00520, map01100, map01110, map01120, map01130, map01200 | Serratia Leuconostoc |
| tpiA | D20vsD40 D20vsD60 | K01803 | map00010, map00051, map00562, map00710, map01100, map01110, map01120, map01130, map01200, map01230 | Serratia Lactococcus |
| eno | D20vsD40 D20vsD60 | K01689 | map00010, map00680, map01100, map01110, map01120, map01130, map01200, map01230, map03018, map04066 | Pseudomonas Leuconostoc |
| alaS | D20vsD40 D20vsD60 | K01652 | map00290, map00650, map00660, map00770, map01100, map01110, map01130, map01210, map01230 | Leuconostoc Rahnella |
| bglA | D40vsD60 | K01223 | map00010, map00500 | Pediococcus |
| tktA | D20 vs D40 | K00615 | map00030, map00710, map01051, map01100, map01110, map01120, map01130, map01200, map01230 | Serratia Pseudomonas |
| gor | D20 vs D60 | K00383 | map00480, map04918 | Pseudomonas Serratia |
| pdhD | D20vsD40 | K00382 | map00010, map00020, map00260, map00280, map00620, map00630, map00640, map01100, map01110, map01120, map01130, map01200 | Lactobacillus Carnobacterium |
| aceE | D20vsD40 | K00163 | map00010, map00020, map00620, map01100, map01110, map01120, map01130, map01200 | Pseudomonas |
| gnd | D40vsD60 | K00033 | map00030, map00480, map01100, map01110, map01120, map01130, map01200 | Pseudomonas |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Yan, H.; Xu, X.; Xiao, X.; Yuan, Y.; Guo, N.; Zhang, T.; Li, M.; Zhu, L.; Zhao, C.; et al. Metagenomic/Metaproteomic Investigation of the Microbiota in Dongbei Suaicai, a Traditional Fermented Chinese Cabbage. Fermentation 2024, 10, 185. https://doi.org/10.3390/fermentation10040185
Zhang Y, Yan H, Xu X, Xiao X, Yuan Y, Guo N, Zhang T, Li M, Zhu L, Zhao C, et al. Metagenomic/Metaproteomic Investigation of the Microbiota in Dongbei Suaicai, a Traditional Fermented Chinese Cabbage. Fermentation. 2024; 10(4):185. https://doi.org/10.3390/fermentation10040185
Chicago/Turabian StyleZhang, Yamin, Haiyang Yan, Xiangxiu Xu, Xiaowei Xiao, Yuan Yuan, Na Guo, Tiehua Zhang, Mengyao Li, Ling Zhu, Changhui Zhao, and et al. 2024. "Metagenomic/Metaproteomic Investigation of the Microbiota in Dongbei Suaicai, a Traditional Fermented Chinese Cabbage" Fermentation 10, no. 4: 185. https://doi.org/10.3390/fermentation10040185