

Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae

1
State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China
2
University of Chinese Academy of Sciences, Beijing 100049, China
*
Authors to whom correspondence should be addressed.
†
Current address: Department of Physiology, Faculty of Basic Medical Sciences, Hubei University of Medicine, Shiyan 442000, China.
Fermentation 2023, 9(10), 886; https://doi.org/10.3390/fermentation9100886
Submission received: 11 September 2023
/
Revised: 27 September 2023
/
Accepted: 28 September 2023
/
Published: 29 September 2023
(This article belongs to the Special Issue Research on Microbial Transformation and Biosynthesis of Enzymes)
Abstract
:As a life-essential coenzyme, nicotinamide adenine dinucleotide (NAD+) has been explored for more than a century. In Saccharomyces, the natural NAD+ de novo biosynthetic pathway initiating from tryptophan has been well elucidated. To bypass this stringently controlled natural pathway in yeast, an economical C3N pathway that was developed in Escherichia coli previously was constructed in Saccharomyces as a short detour for de novo NAD+ biosynthesis. After the functional expressions of the C3N genes were identified in Saccharomyces cerevisiae BY4741 by in vitro enzymatic assays, the C3N module was introduced into an NAD+ auxotrophic S. cerevisiae strain BY01, in which the BNA2 gene encoding tryptophan 2,3-dioxygenase was inactivated. The efficient NAD+ synthesis via the C3N pathway was confirmed by both plate assays and fermentation analysis. The applicability of the C3N pathway in cofactor engineering was tested by introducing it into S. cerevisiae BY4741, which improved the cellular NAD(H) level considerably. Consequently, this study proved that the de novo NAD+ biosynthetic pathway can be replaced by an artificial pathway in yeast, which paves a way to design more promising schemes in eukaryotes for rational manipulation of the cellular NAD(H) levels.
1. Introduction
Nicotinamide adenine dinucleotide (NAD+) and its reduced form NADH predominately function as electronic donor and acceptor in more than a quarter of the cellular redox reactions [1]. In addition, NAD+ is a substrate of many NAD+-consuming enzymes, which play essential roles in signal transduction pathways regulating crucial biological processes, e.g., DNA repair, transcription, and cell cycle progression [2]. Apart from the important physiological functions of NAD+, a common cognition about NAD+ was its cellular supply. Engineering to modulate the cellular concentration of NAD(H) is a practical way to tune the catalytic efficiencies of NAD(H)-dependent and NAD+-consuming enzymes, which has been performed not only for research purposes in prokaryotic and eukaryotic cells but also for valuable products bio-manufacturing in industry [3,4]. Several different strategies have been developed to modulate cellular NAD(H) levels, including supplementation of NAD+ or its precursors, reduction of NAD+ consumption, and reinforcement of NAD+ biosynthesis pathways [5,6]. The efficiency of the last strategy has been proven in many cases; people can expand the cellular NAD(H) pools and accelerate its replenishment by strengthening the de novo biosynthesis pathway, as well as the salvage pathway of NAD+ [7,8].
To date, only two natural de novo NAD+ biosynthesis pathways have been delineated [4,9]. Pathway I, which distributes in plants and most bacteria, uses l-aspartate (l-Asp) as a precursor for the nicotinamide moiety of NAD+ [10,11], whereas pathway II, which exists in some bacteria, mammals, and fungi, recruits l-tryptophan (l-Trp) as a precursor [4,12]. The de novo biosynthesis pathways of NAD+ are essential to cells in most cases and are tightly controlled at transcriptional, translational, and post-translational levels [13,14,15], which may explain the difficulties in modulating cellular NAD(H) levels by engineering on those pathways. Recently, we constructed an alternative de novo NAD+ biosynthesis pathway in Escherichia coli, the C3N pathway, which uses chorismate as a starter unit and converts it to 3-hydroxyanthranilic acid (3-HAA) by three enzymes, PhzD, PhzE, and DhbX, from secondary metabolism [16]. Subsequently, 3-HAA is converted to quinolinic acid (QA) by 3-HAA 3,4-dioxygenase, which is also a key enzyme in pathway II for NAD+ synthesis [17]. The three steps from QA to NAD+ are conserved for all de novo biosynthesis pathways of NAD+ [9]. The newly developed C3N pathway circumvents the stringent regulation on de novo NAD+ biosynthesis and results in a more than nine times improvement of the cellular NAD(H) level in E. coli, demonstrating its powerfulness in cofactor engineering [16]. However, all enzymes used for constructing the C3N pathway in E. coli are of bacterial origins; the applicability of this pathway in eukaryotic cells remains to be exploited.
As a classical representative of eukaryotic cells, Saccharomyces cerevisiae is a generally recognized as safe (GRAS) industrialized cell for the production of valuable chemicals, such as food additives, natural products, and pharmaceutical precursors [18]. It is also an ideal molecular biology model for the study of NAD+-dependent physiological processes such as aging and related diseases and signaling pathways [19,20]. S. cerevisiae synthesizes NAD+ from l-Trp via de novo pathway II [21]. As depicted in Figure 1, l-Trp is converted to 3-HAA, the shared intermediate of pathway II and the C3N pathway, by four steps. Moreover, l-Trp is derived from chorismate by five enzymatic reactions. Therefore, it takes nine steps for the conversion of chorismate to 3-HAA during the native NAD+ biosynthesis in S. cerevisiae. The C3N pathway only needs three steps for the conversion of chorismate to 3-HAA, which is certainly an economical short detour for NAD+ biosynthesis. Actually, almost all reported successful cases about improving the cellular NAD(H) levels in S. cerevisiae are via manipulations on the salvage pathway instead of the de novo pathway, which may reveal the difficulty in modulating the tightly controlled pathway II for cofactor engineering [21]. The artificial C3N pathway may circumvent the native regulation and serves as an alternative for expanding the cellular NAD(H) pool of S. cerevisiae. In addition, the C3N pathway decouples protein synthesis and NAD+ de novo biosynthesis by recruiting a non-proteinogenic amino acid precursor, which makes it suitable for biotransformation that needs protein overexpression.
In this work, we firstly verified that all three necessary bacterial genes (forming the C3N module) for the construction of C3N pathway could be functionally expressed in S. cerevisiae. An NAD+ auxotrophic S. cerevisiae strain that could survive on NAD+ synthesized solely by the C3N pathway was then constructed, revealing that the C3N pathway is also efficient in eukaryotes. When the C3N module was expressed in wild-type S. cerevisiae, a considerable increment of the cellular NAD(H) level was observed, demonstrating the applicable potential of the C3N module in cofactor engineering.
2. Materials and Methods
2.1. Strains, Plasmids, and Growth Conditions
Strains and plasmids used in this study were summarized in Table S1. E. coli JM109 was used for DNA cloning and propagation of plasmids. S. cerevisiae BY4741 was used for expressing recombinant proteins. LB medium (10 g/L peptone, 5 g/L yeast extract, and 10 g/L NaCl) with or without 20 g/L agar was used for the growth of E. coli strains and propagation of plasmids at 37 °C, and, if necessary, 100 μg/mL ampicillin and 50 μg/mL kanamycin were added. YPD medium (10 g/L yeast extract, 20 g/L peptone, and 20 g/L glucose), nicotinic acid drop-out YMM medium (YMMN medium) [22], and yeast nitrogen base supplemented with essential nutrients medium (YNB-S) (6.7 g/L Yeast nitrogen base without amino acids (#Y0626, Sigma, Beijing, China), 1.92 g/L Yeast Synthetic Drop-out Media Supplements (#Y1501, Sigma, Beijing, China), and 20 g/L glucose) with or without 20 g/L agar were used for growth of S. cerevisiae strains at 30 °C. If necessary, 150 mg/L uracil and/or 1 g/L 5-fluoroorotic acid were supplemented.
2.2. General DNA Manipulations and Sequence Analyses
DNA synthesis and sequencing were performed by Generay Biotech (Shanghai, China) and Genewiz (Suzhou, China), respectively. The classical lithium acetate/single-stranded carrier DNA/PEG method was used for S. cerevisiae genetic manipulation and plasmid transformation according to the standard protocols [23]. The chemical transformation method was used for E. coli transformation and plasmids propagation according to the standard protocols [24]. To construct the different engineered plasmids, the one-step cloning method was employed according to the manufacturer’s instructions (Vazyme, Nanjing, China). PCRs were performed with the Q5 DNA polymerase (NEB, Beijing, China). The digestion of plasmids was carried out by restriction enzymes (Takara, Dalian, China) following the general methods [25].
2.3. Construction of S. cerevisiae BY01
To inactivate the tryptophan 2,3-dioxygenase gene BNA2, the about 0.5 kb upstream and downstream flanked fragments of BNA2 were amplified with primers BNA2-U-F (TTTGCCCTCCAATTCCCTAGGGATTGGCTAACGATGGTTGG)/BNA2-U-D (TGGCGTAATAGCGAAGAGGCCATCGGCGTTGACTCTTTCTT) and BNA2-D-F (CAAAAGCTGGAGCTGGCCTTGTGATAGACGGGTTGGAGGGC)/BNA2-D-D (CATCGTTAGCCAATCCCTAGGGAATTGGAGGGCAAAATAAT) from the S. cerevisiae BY4741 genome and inserted into the SfiI site of pUMRI-21(A) to generate pRU-BNA-UD, which was then linearized by AvrII and introduced into S. cerevisiae BY4741. The transformants were spread on YNB-S plates without uracil and cultured at 30 °C for 3 days. One of the transformants was inoculated into 10 mL YPD medium and cultured at 30 °C, 220 rpm for 24 h. The cells of 200 μL culture were collected by centrifugation, washed twice with ddH2O, and spread on YNB-S plates with 150 mg/L uracil and 1 g/L 5-fluoroorotic acid. After incubation at 30 °C for 3 days, one of the transformants was verified as BY01 by primers BNA2-F (GATTGGCTAACGATGGTTG)/BNA2-R (GAATTGGAGGGCAAAATAAT).
2.4. Expression of phzD, phzE, and dhbX Individually in S. cerevisiae
To express phzD in S. cerevisiae BY4741, the codon-optimized phzD gene was synthesized and inserted into plasmid pYES2.0 to afford pYES2.0-phzD. The 0.4 kb TEF1 promoter PTEF1 was cloned from the S. cerevisiae genome by primer pair TEF-DR (GCTTGGTACCGAGCTCGGATCGATCTTCAAAATGTTTCTAC)/TEF1p-R (ATAGTGAGTCGTATTACGGATCCTAGAAAACTTAGATTAGA) and was ligated with the 6.1 kb PCR fragment amplified with primer pairs Gal-OF (GAGCTCGGTACCAAGCTACTAGTGGATCATCCCCACG)/T7 (TAATACGACTCACTATAGG) using pYES2.0-phzD as a template to construct pYE-PTEF1-phzD, in which the GAL1 promoter PGAL1 was replaced by the promoter PTEF1. S. cerevisiae BY00-phzD was then obtained by introducing pYE-PTEF1-phzD into S. cerevisiae BY4741 by chemical transformation. As described above, constructions of S. cerevisiae BY00-phzE and BY00-dhbX were carried out using the same procedures. To express the proteins PhzD, PhzE, and DhbX individually in S. cerevisiae, the engineered strain was inoculated in YNB-S medium without uracil and cultured at 30 °C, 220 rpm for 48 h. The seeds culture was inoculated into YPD medium at 1% ratio (v/v) and cultured at 30 °C, 220 rpm for 24–36 h. The cells were harvested by centrifugation (4 °C, 3000× g, 20 min) and re-suspended in lysis buffer (20 mM Tris-HCl, 500 mM NaCl, 5 mM imidazole, pH 7.9) and burst by ultrasonication, and the cell debris was removed by centrifugation (4 °C, 16,000× g, 30 min). The crude proteins PhzD, PhzE, and DhbX were desalted and concentrated by ultrafiltration using a Millipore centrifugal filter (15 mL, 10 kD MWCO), respectively, and stored at −80 °C in HEPES buffer (pH 8.0, for PhzD and PhzE) and in PBS buffer (pH 7.4, for DhbX) with 20% glycerol [26], respectively.
Functions of those three proteins were verified by enzymatic assays using the crude proteins as catalysts. The PhzE activity was tested in a 50 μL mixture containing 100 mM HEPES buffer (pH 8.0), 1 mM chorismate, 10 mM glutamine, 10 mM MgCl2, and 20 μL crude PhzE at 30 °C for 1 h; the PhzD activity was checked in a 50 μL mixture containing 100 mM HEPES buffer (pH 8.0), 1 mM chorismate, 10 mM glutamine, 10 mM MgCl2, 5 μL crude PhzD, and 20 μL crude PhzE at 30 °C for 1 h; the DhbX activity was detected in a 50 μL mixture containing 50 mM PBS buffer (pH 7.4), 2 mM NAD+, 2 mM DHHA, and 10 μM crude DhbX at 37 °C for 1 h. BY4741 with the empty pYES2.0 plasmid (designated as S. cerevisiae BY00-Con) was used as a control. The reactions were quenched with an equal volume of CHCl3. After centrifugation, the aqueous phases of the reactions (10 μL) were analyzed by HPLC.
2.5. Construction of S. cerevisiae BY00-Con, BY00-C3N, BY01-Con, and BY01-C3N
To construct S. cerevisiae BY01-C3N, the 0.6 kb phzD gene was PCR cloned from pYES2.0-phzD with primer pair phzD-F (ATGCATCATCATCATCATCATTCAGGTATTCCAG)/phzD-R (CGAAGAATTGTTAATTAAGAGCTCTTATTCCAAGACTTC) and inserted into the SacI/EcoRI sites of pUMRI-21(A) to generate pRU-ZD (Ga). The about 1.9 kb phzE gene was amplified from pYES2.0-phzE using primer pair phzE-F (CTCGAGTTAAGGTCTTCTTTCAACCA)/phzE-R (GTAATACGACTCACTATAGGGTCGACATGCATCATCATCATCATCATAACGC) and inserted into the SaII/XhoI sites of pRU-ZD (Ga) to generate pRU-ZDE (Ga). Then, the 0.4 kb TEF1 promoter PTEF1 and 0.6 kb PGK1 promoter PPGK1 were amplified using S. cerevisiae genome as a template with primer pair TEF1P-F (GTTTGCCCCTACGTTTTGCGATCTTCAAAATGTTTCTA)/TEF1P-R (ATAGTGAGTCGTATTACGGATCCTAGAAAACTTAGATTAGA) and PGK1-F (GCAAAACGTAGGGGCAAACCCGATTTGGGCGCGAATCC)/PGK1-R (GAATGATGATGATGATGATGCATTGTTTTATATTTGTTGTAAAAAG), respectively. The two promoters were inserted upstream of phzE and phzD in pRU-ZD (Ga), respectively, using the one-step cloning method to generate pRU-ZDE (Co). The 4.2 kb cassette (TCYC1-phzE-PTEF1-PPGK1-phzD-TTDH1) was amplified from pRU-ZDE (Co) using primer pair phzDE-F (CGCGTGGGGATGATCCACTAGTAGCTTGGTACCCTTCGAGCGTC)/phzDE-R (CGATCCGAGCTCGGTACCGGCCTCTTCGCTATTACGCC) and inserted into the KpnI site of pYE-PTEF1-dhbX to generate pY-C3N. S. cerevisiae BY01-C3N and BY00-C3N were then obtained by introducing pY-C3N into S. cerevisiae BY01 and BY00, respectively. The control strain S. cerevisiae BY00-Con and BY01-Con was constructed by introducing empty pYES2.0 into S. cerevisiae BY00 and BY01, respectively.
2.6. Growth of S. cerevisiae Strains on YMMN Plates
The different S. cerevisiae strains were inoculated into 3 mL YNB-S medium (with or without 150 mg/L uracil) for 48 h. The seeds were inoculated into 10 mL YMMN medium (with or without 150 mg/L uracil) and cultured for 24 h. The cells were then collected by centrifugation and washed twice by ddH2O. For the growth experiments on agar plate, 1.5 μL dilutions (OD600 of 0.1) of different strains were spotted onto YMMN agar and grown at 30 °C with varied supplements. S. cerevisiae BY00 was spotted onto YMMN agar supplemented with 150 mg/L uracil; S. cerevisiae BY01 was inoculated onto YMMN agar (150 mg/L uracil) with or without 10 mM QA; S. cerevisiae BY01-Con was inoculated onto YMMN agar with or without 10 mM QA; and S. cerevisiae BY01-C3N was spotted onto YMMN agar directly.
2.7. Measurement of Intracellular NAD(H) Concentrations
The different S. cerevisiae strains were inoculated into 3 mL YNB-S medium (with or without 150 mg/L uracil) for seed growth under 30 °C, 220 rpm for 48 h. The cells were collected by centrifugation, washed twice with ddH2O, and resuspended into YMMN medium. Then, the growth condition of the engineering strains was first tested by test tube with 3 mL YMMN medium; subsequently, the different strains were cultured into 20 mL YMMN medium in a 100 mL shake flask at a final OD600 of 0.02. The culture was grown at 30 °C, 220 rpm for 36 h. To measure the intracellular NAD(H) concentration, the cells were harvested by centrifugation (4 °C, 3000× g, and 5 min) at the time point of four hours after entering the stationary phase. The cell pellets were washed with ice-cold PBS buffer, re-suspended in the same buffer, and adjusted to OD600 of 1.0. The concentrations of NAD(H) (total NAD+ and NADH) and NAD+ only were measured by enzyme cycling-based colorimetric assay [27]. S. cerevisiae was calculated as 3 × 107 cells/mL at OD600 of 1.0, and the volume of one S. cerevisiae cell was 70 µm3 [28,29]. The growth curve was supervised by Multi-mode Microplate Reader (Synergy H1, BioTek, Winooski, VT, USA) with an interval of 6 h.
2.8. Spectroscopic Analysis
HPLC analysis was performed with a ZORBAX SB-Aq Stable-Bond Analytical column (5 μm, 4.6 × 250 mm, Agilent Technologies, Santa Clara, CA, USA) on a Shimadzu HPLC system (Shimadzu, Kyoto, Japan) [30]. The column was developed with solvent A (water with 0.1% trifluoroacetic acid) and acetonitrile at a flow rate of 1 mL/min except for the DHHA dehydrogenase assays (0.8 mL/min). For analysis of the DHHA dehydrogenase reactions, the percentage of acetonitrile was changed linearly from 1% to 50% over 0–30 min, from 50% to 100% over 30–32 min, and maintained at 100% for 5 min; for analysis of the crude PhzD- and/or PhzE-containing enzymatic assays, the ratio of acetonitrile was changed linearly from 0% to 5% over 0–2 min, from 5% to 50% over 2–17 min, from 50% to 100% over 19–21 min, and maintained at 100% for 10 min. To detect the intermediates of NAD+ biosynthesis in S. cerevisiae strains, the ratio of acetonitrile was initially stayed 0% over 1 min, changed from 0% to 5% over 1–4 min, from 5% to 20% over 4–19 min, from 20% to 22% over 19–21 min, from 22% to 100% over 21–23 min, and maintained at 100% for 10 min. The detection wavelength of chorismate, ADIC, and DHHA was 280 nm, and it was 294 nm for 3-HAA analysis.
2.9. Sequence Data
The GenBank accession numbers of the original phzE, phzD, and dhbX genes are AAC64488, AAC64487, and CDG76955, respectively. The GenBank accession numbers of the corresponding codon-optimized phzE, phzD and dhbX genes are OK275489, OK275488, and OK275490, respectively.
3. Results
3.1. Functional Expression of the C3N Pathway Genes in S. cerevisiae
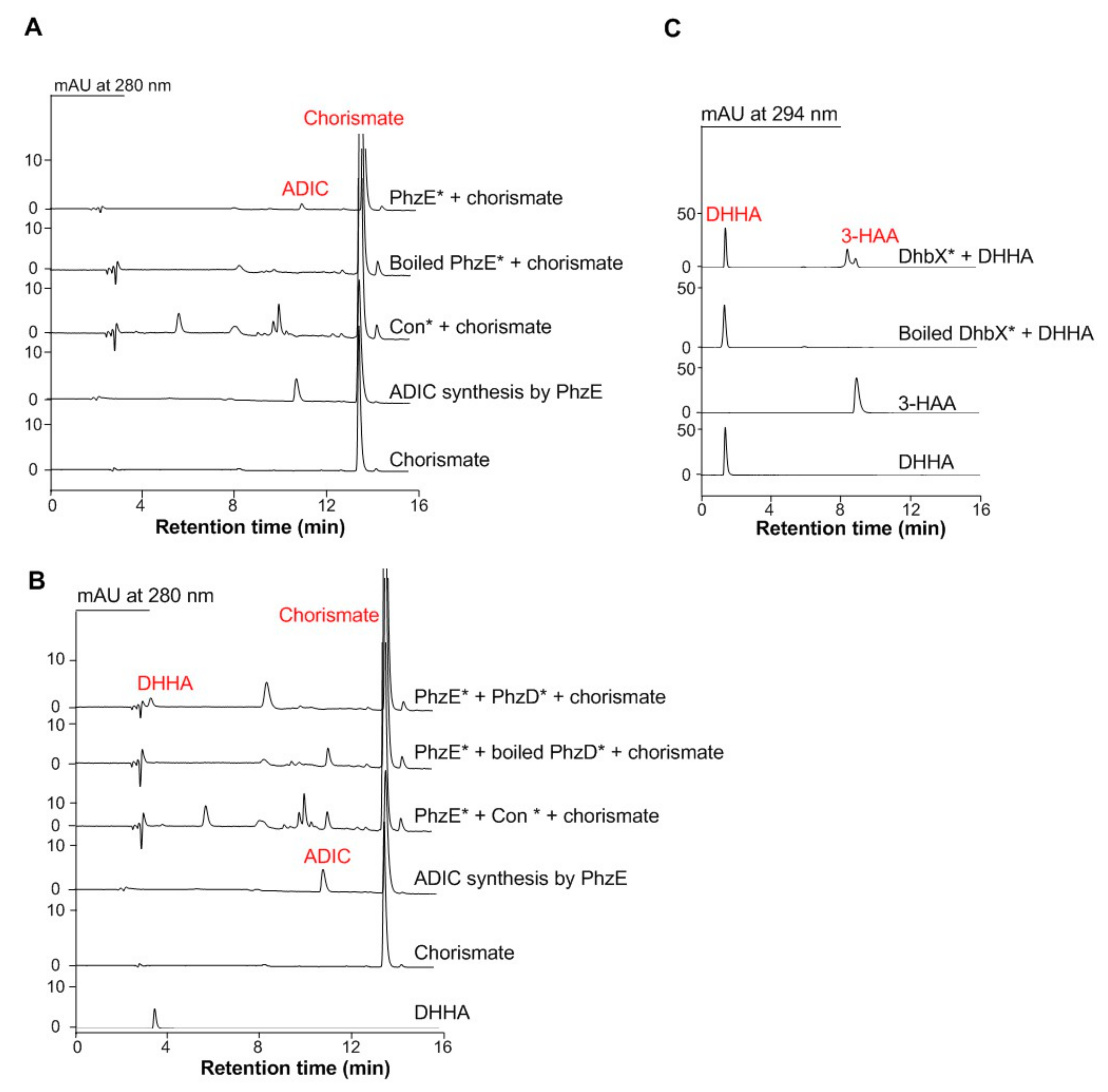
As aforementioned, three enzymes that can convert chorismate to 3-HAA are needed for building the short detour NAD biosynthesis pathway in S. cerevisiae. The genes that encode the three enzymes of 2-amino-2-deoxyisochorismate (ADIC) synthase PhzE, 2,3-dihydro-3-hydroxyanthranilic acid (DHHA) synthase PhzD, and DHHA dehydrogenase DhbX are all from Gram-positive bacteria with high G + C genomes [31,32]. Therefore, the codon-optimized phzE, phzD, and dhbX genes were synthesized and inserted into pYES2.0 plasmid under the control of the constitutive TEF1 promoter PTEF1, and then introduced into wild S. cerevisiae BY4741 (BY00) to construct the individual gene expression strain S. cerevisiae BY00-phzE, BY00-phzD, and BY00-dhbX, respectively. The functions of those proteins were further verified by in vitro enzymatic reactions. It was shown that the crude proteins of S. cerevisiae BY00-phzE could convert chorismate to ADIC (Figure 2A); the mixture crude proteins of S. cerevisiae BY00-phzE and S. cerevisiae BY00-phzD could convert chorismate to DHHA together (Figure 2B), and the protein of S. cerevisiae BY00-dhbX could convert DHHA to 3-HAA (Figure 2C). Overall, the results suggested that the phzE, phzD, and dhbX genes could be used to build the shorter C3N pathway in S. cerevisiae to replace the native de novo NAD+ biosynthesis pathway II.
3.2. Construction of the C3N Pathway in S. cerevisiae
To test the applicability of C3N pathway in S. cerevisiae, we constructed an NAD+ auxotrophic S. cerevisiae strain BY01 by knocking out the BNA2 gene, which encodes a tryptophan 2,3-dioxygenase that catalyzes the first step of de novo NAD+ biosynthesis pathway II [33] (Figure 1 and Figure 3A). As 3-HAA is not stable and tends to be oxidized in air, we used QA as a supplement and added to the YMMN solid medium for growth experiment. As anticipated, the wild-type strain S. cerevisiae BY00 could grow well on YMMN plate, whereas the NAD+ auxotrophic strain S. cerevisiae BY01 was unable to grow on YMMN plate unless QA was supplemented (Figure 3B). It suggested that the pathway converting l-Trp to 3-HAA was blocked, whereas the enzymes responsible for the late steps of pathway II that converts 3-HAA to NAD+ are functionally expressed in S. cerevisiae BY01.
Subsequently, we constructed pY-C3N by sequentially cloning phzE and phzD into plasmid pYE-PTEF1-dhbX and transformed the resultant plasmid pY-C3N into S. cerevisiae BY01 to generate S. cerevisiae BY01-C3N with a complete C3N pathway. A control strain S. cerevisiae BY01-Con was constructed by transforming the empty pYES2.0 plasmid into S. cerevisiae BY01. As anticipated, S. cerevisiae BY01, S. cerevisiae BY01-Con was unable to grow on YMMN plate without QA supplementation (Figure 3B). In contrast, S. cerevisiae BY01-C3N could grow well on YMMN plate without QA supplementation, indicating that the C3N pathway was successfully constructed in BY01-C3N and could synthesize NAD+ efficiently for its growth.
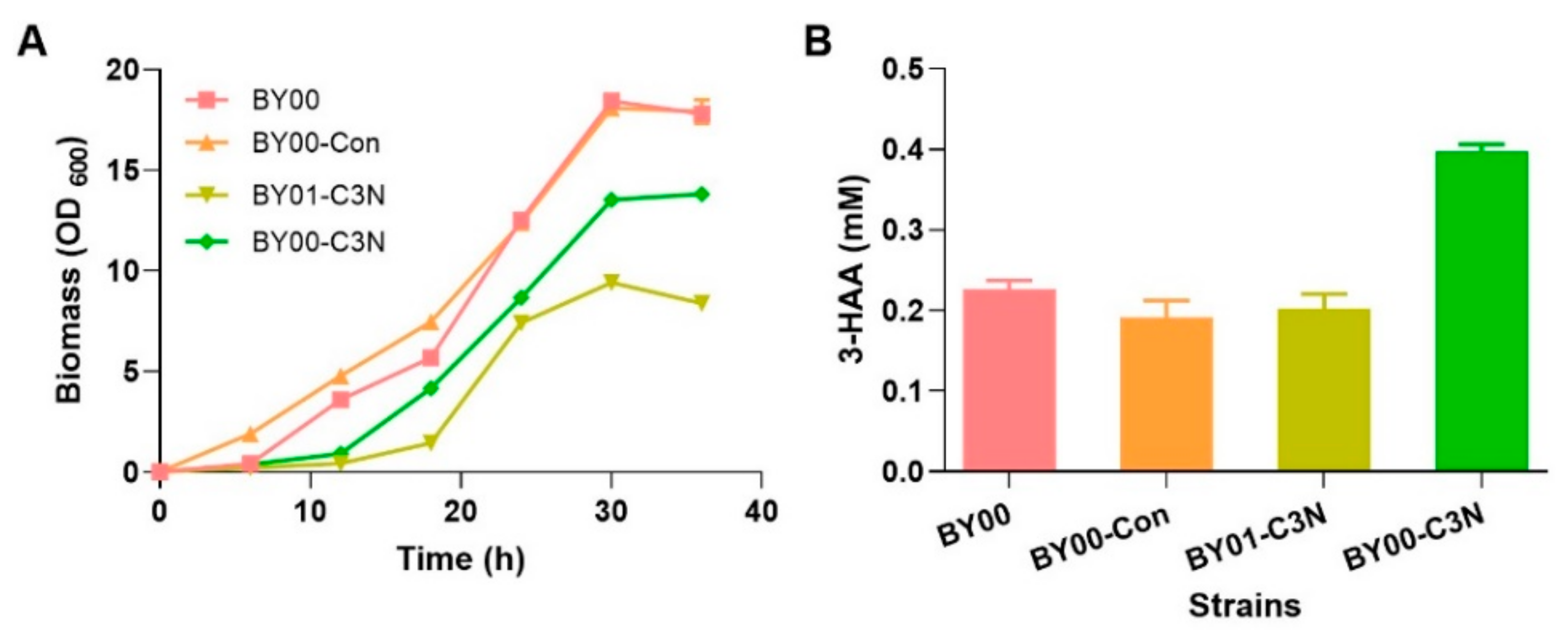
Subsequently, S. cerevisiae BY00, BY00-Con, BY01, BY01-Con, and BY01-C3N were cultivated in YMMN liquid medium for evaluating their growth. As anticipated, although slightly vague colony formed on solid plate with an initial OD600 = 0.1, S. cerevisiae BY01 and BY01-Con were unable to grow in liquid YMMN medium without QA supplementation, as shown in Figure S1. Meanwhile, compared to the BY00 and BY00-Con exhibited normal growth curves, the mutant strain BY01-C3N displayed a clear growth delay (Figure 4A). Cellular NAD(H) levels of those strains were measured when the engineered strains reached early stationary phase. The cellular NAD(H) level of BY00-Con (2.92 ± 0.20 mM) was slightly lower than that of BY00 (3.30 ± 0.45 mM) (Table 1), indicating that the introduction of pYES2.0 had some influence on cell metabolism. The cellular NAD(H) level of BY01-C3N (3.13 ± 0.06 mM) was comparable to that of BY00 (Table 1), implying that the constructed C3N pathway is as efficient as the native de novo NAD+ biosynthesis pathway II in S. cerevisiae.
3.3. Expanding the NAD(H) Pool of S. cerevisiae by the C3N Pathway
To further explore cofactor engineering potency of the C3N pathway in S. cerevisiae, strain BY00-C3N was constructed by introducing the C3N module into S. cerevisiae BY00. In BY00-C3N, the key intermediate 3-HAA for NAD+ biosynthesis could be synthesized via both the C3N pathway that starts from chorismate and pathway II that starts from l-Trp (Figure 1). When cultivated in YMMN liquid medium, BY00-C3N displayed a similar growth deficiency phenomenon as BY01-C3N (Figure 4A), which may be caused by the metabolic burden of the C3N module overexpression. Analysis of the cellular metabolites revealed that an excess amount of 3-HAA (0.40 ± 0.01 mM) was accumulated in BY00-C3N compared to the other three S. cerevisiae strains (BY00, BY00-Con, and BY01-C3N), in which 3-HAA was accumulated at nearly a half level (Figure 4B), no other metabolites were identified by HPLC. The cellular NAD(H) level of BY00-C3N reached 4.59 ± 0.37 mM, which was about 40% higher than that of BY00 (Table 1), suggesting that the C3N pathway has a potential to elevate the cellular level of NAD(H) in S. cerevisiae.
4. Discussion
NAD+ is a life-essential metabolite involved in numerous redox reactions and multiple cellular physiological processes. Since it was discovered as the first ‘cozymase’ more than 100 years ago, two natural de novo NAD+ biosynthetic pathways (pathway I and II) have been elucidated and both of them start from a proteinogenic amino acid [4,9]. In a previous study, we designed an alternative de novo NAD+ biosynthesis pathway, the C3N pathway that starts from chorismate, and established this pathway successfully in E. coli by expressing four exogenous genes encoding an ADIC synthase, a DHHA synthase, a DHHA dehydrogenase, and a 3-HAA 3,4-dioxygenase [16]. Chorismate can be converted to 3-HAA by the former three enzymes, and then undergo the oxidative ring cleavage catalyzed by 3-HAA 3,4-dioxygenase and a spontaneous cyclization to form QA, which will enter the native de novo NAD+ biosynthetic pathway I of E. coli and generate NAD+ via three steps common to all de novo NAD+ biosynthetic pathways [9,16]. Our previous work demonstrated the advantages of the C3N pathway in prokaryotic cells. In this study, we showed that the C3N pathway is also applicable in eukaryotic cells using S. cerevisiae as a model system. S. cerevisiae synthesizes NAD+ via de novo NAD+ biosynthetic pathway II, which also recruits 3-HAA 3,4-dioxygenase to convert 3-HAA to QA [17]. Therefore, only ADIC synthase, DHHA synthase, and DHHA dehydrogenase are necessary to build the C3N pathway in S. cerevisiae. The biosynthesis of NAD+ in S. cerevisiae BY01, a mutant strain deficient in synthesizing 3-HAA, could be restored to a normal level by introducing the C3N module, presenting the first example of eukaryotic cells that can survive on the artificial C3N pathway. Compared to the native chorismate to 3-HAA process composed of nine steps in yeast, only three steps are needed in the short detour of the C3N pathway [21].
It was shown that the C3N pathway could increase the cellular NAD(H) concentration more than nine times to about 9.3 mM in E. coli [16]. To explore the cofactor engineering potency of the C3N pathway in S. cerevisiae, the genes of C3N pathway were introduced to S. cerevisiae wild-type strain to generate BY00-C3N, in which the native de novo NAD+ biosynthesis pathway II is effective, and the build-in C3N module will supply extra amount of 3-HAA. As anticipated, the cellular NAD(H) level of BY00-C3N was considerably higher than that of BY00-Con or BY00, identifying its applicability in cofactor engineering in eukaryotic cells. However, it was not as effective as using the C3N module for cellular NAD(H) level improvement in E. coli. One possible reason is that the three C3N module enzymes from bacteria were not expressed well in S. cerevisiae. As shown in Figure 2, we could observe a poorly catalytic ability of the enzymes PhzE and PhzD expressed into S. cerevisiae. Meanwhile, the naturally competing metabolic pathways of chorismate may be other reasons that caused the ineffectiveness of the C3N pathway. Moreover, metabolic profile analysis of BY00-C3N revealed that more 3-HAA was accumulated compared to the other engineered strains, revealing that the 3-HAA 3,4-dioxygenase from pathway II could not convert the extra 3-HAA synthesized by the C3N module to QA efficiently [21]. Those work pave a way for further improvement of the cellular NAD(H) levels in yeast.
As a product of the shikimate pathway, chorismate is a natural branch point for various metabolic pathways [34,35,36]. It is abundant in many different cells including bacteria, archaea, fungi, and plants [37]. Therefore, the C3N pathway has the potential to be used in diverse organisms. Here, we showed that, in addition to the prokaryotic E. coli cells, the C3N pathway could supply NAD+ efficiently in the eukaryotic yeast cells via a more economical short detour, which set the stage for the application of this pathway in the other eukaryotes like fungi and plants.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/fermentation9100886/s1, Figure S1: The growth phenotypes of different engineered S. cerevisiae in YMMN medium. Table S1: Strains and plasmids used in this study.
Author Contributions
Conceptualization, X.L. and Y.C.; methodology, X.L., Y.T. and Y.D.; validation, X.L., Y.T. and P.L.; investigation, X.L. and Y.D.; Writing—Original draft preparation, X.L.; Writing—Review and editing, X.L., Y.T. and Y.C.; supervision, P.L. and Y.C.; funding acquisition, Y.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by in part by the National Key R&D Program of China (2018YFA0901600), the National Natural Science Foundation of China (32370058 and 32025002), and the Transformation Program of S&T Achievements of Inner Mongolia (2020CG0012).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no competing interests.
References
- Selles, V.L.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Et Biophys. Acta (BBA) Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, B.; Fang, Y.; Tan, T. Cofactor engineering for more efficient production of chemicals and biofuels. Biotechnol. Adv. 2017, 35, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2018, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.J.; Yang, W.; Wang, L.; Zhu, Z.; Zhang, S.; Zhao, Z.K. Engineering NAD+ availability for Escherichia coli whole-cell biocatalysis: A case study for dihydroxyacetone production. Microb. Cell Factories 2013, 12, 103. [Google Scholar] [CrossRef]
- Li, F.; Li, Y.; Cao, Y.; Wang, L.; Liu, C.; Shi, L.; Song, H. Modular engineering to increase intracellular NAD(H/+) promotes rate of extracellular electron transfer of Shewanella oneidensis. Nat. Commun. 2018, 9, 3637. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Zhao, G.; Caiyin, Q.; Qiao, J. Redox cofactor engineering in industrial microorganisms: Strategies, recent applications and future directions. J. Ind. Microbiol. Biotechnol. 2018, 45, 313–327. [Google Scholar] [CrossRef]
- Sharma, S.; Hsieh, Y.-C.; Dietze, J.; Bockwoldt, M.; Strømland, Ø.; Ziegler, M.; Heiland, I. Early Evolutionary Selection of NAD Biosynthesis Pathway in Bacteria. Metabolites 2022, 12, 569. [Google Scholar] [CrossRef]
- Heuser, F.; Schroer, K.; Lütz, S.; Bringer-Meyer, S.; Sahm, H. Enhancement of the NAD(P)H pool in Escherichia coli for biotransformation. Eng. Life Sci. 2007, 7, 343–353. [Google Scholar] [CrossRef]
- Katoh, A.; Uenohara, K.; Akita, M.; Hashimoto, T. Early steps in the biosynthesis of NAD+ in Arabidopsis start with aspartate and occur in the plastid. Plant Physiol. 2006, 141, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Kurnasov, O.; Goral, V.; Colabroy, K.; Gerdes, S.; Anantha, S.; Osterman, A.; Begley, T.P. NAD biosynthesis: Identification of the tryptophan to quinolinate pathway in bacteria. Chem. Biol. 2003, 10, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.A.; De Ingeniis, J.; Mancini, C.; Cimadamore, F.; Zhang, H.; Osterman, A.L.; Raffaelli, N. Transcriptional regulation of NAD+ metabolism in bacteria: NrtR family of Nudix-related regulators. Nucleic Acids Res. 2008, 36, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Malkowski, S.N.; Spencer, T.C.J.; Breaker, R.R. Evidence that the nadA motif is a bacterial riboswitch for the ubiquitous enzyme cofactor NAD+. RNA 2019, 25, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Croft, T.; Venkatakrishnan, P.; Raj, C.J.T.; Groth, B.; Cater, T.; Salemi, M.R.; Phinney, B.; Lin, S.J. N-terminal protein acetylation by NatB modulates the levels of Nmnats, the NAD+ biosynthetic enzymes in Saccharomyces cerevisiae. J. Biol. Chem. 2020, 295, 7362–7375. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, X.L.; Horsman, G.P.; Li, P.W.; Wang, M.; Li, J.E.; Zhang, Z.; Liu, W.; Wu, B.; Tao, Y.; et al. Construction of an alternative NAD+ de novo biosynthesis pathway. Adv. Sci. 2021, 8, 2004632. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, K.; Yang, Y.; Davis, I.; Liu, A. Observing 3-hydroxyanthranilate-3,4-dioxygenase in action through a crystalline lens. Proc. Natl. Acad. Sci. USA 2020, 117, 19720–19730. [Google Scholar] [CrossRef]
- Hammer, S.K.; Avalos, J.L. Harnessing yeast organelles for metabolic engineering. Nat. Chem. Biol. 2017, 13, 823–832. [Google Scholar] [CrossRef]
- Chen, R.E.; Thorner, J. Function and regulation in MAPK signaling pathways: Lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef]
- Gershon, H. Gershon, D. The budding yeast, Saccharomyces cerevisiae, as a model for aging research: A critical review. Mech. Ageing Dev. 2000, 120, 1–22. [Google Scholar] [CrossRef]
- Raj, C.J.T.; Lin, S.J. Cross-talk in NAD+ metabolism: Insights from Saccharomyces cerevisiae. Curr. Genet. 2019, 65, 1113–1119. [Google Scholar]
- Oliveira, A.P.; Ludwig, C.; Zampieri, M.; Weisser, H.; Aebersold, R.; Sauer, U. Dynamic phosphoproteomics reveals TORC1-dependent regulation of yeast nucleotide and amino acid biosynthesis. Sci. Signal. 2015, 8, rs4. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2017, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Nojima, H.; Okayama, H. High efficiency transformation of Escherichia coli with plasmids. Gene 1990, 96, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Xie, Z.; Wang, M.; Liu, L.; Zhang, Y.; Wang, W.; Zhang, Z.; Zhao, X.; Li, P.; Guo, Z.; et al. An anaerobic bacterium host system for heterologous expression of natural product biosynthetic gene clusters. Nat. Commun. 2019, 10, 3665. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, M.; Tang, W.; Guo, Z.; Zhang, Y.; Wang, M.; Horsman, G.P.; Zhong, J.; Lu, Z.; Chen, Y. Initiating polyketide biosynthesis by on-line methyl esterification. Nat. Commun. 2021, 12, 4499. [Google Scholar] [CrossRef]
- Kern, S.E.; Price-Whelan, A.; Newman, D.K. Extraction and measurement of NAD(P)+ and NAD(P)H. In Pseudomonas Methods and Protocols; Filloux, A., Ramos, J.L., Eds.; Humana Press: New York, NY, USA, 2014; pp. 311–323. [Google Scholar]
- Day, A.; Schneider, C.; Schneider, B.L. Yeast Cell Synchronization. In Cell Cycle Checkpoint Control Protocols; Lieberman, H., Ed.; Humana Press: New York, NY, USA, 2004; pp. 55–76. [Google Scholar]
- Sporty, J.; Lin, S.J.; Kato, M.; Ognibene, T.; Stewart, B.; Turteltaub, K.; Bench, G. Quantitation of NAD+ biosynthesis from the salvage pathway in Saccharomyces cerevisiae. Yeast 2009, 26, 363–369. [Google Scholar] [CrossRef]
- Tang, W.; Guo, Z.; Cao, Z.; Wang, M.; Li, P.; Meng, X.; Zhao, X.; Xie, Z.; Wang, W.; Zhou, A.; et al. d-Sedoheptulose-7-phosphate is a common precursor for the heptoses of septacidin and hygromycin B. Proc. Natl. Acad. Sci. USA 2018, 115, 2818–2823. [Google Scholar] [CrossRef]
- Culbertson, J.E.; Toney, M.D. Expression and characterization of PhzE from P. aeruginosa PAO1: Aminodeoxyisochorismate synthase involved in pyocyanin and phenazine-1-carboxylate production. Biochim. Biophys. Acta 2013, 183, 240–246. [Google Scholar] [CrossRef]
- Parsons, J.F.; Calabrese, K.; Eisenstein, E.; Ladner, J.E. Structure and mechanism of Pseudomonas aeruginosa PhzD, an isochorismatase from the phenazine biosynthetic pathway. Biochemistry 2003, 42, 5684–5693. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Lee, I.S.; Tsubaki, M.; Kido, R. Tryptophan 2,3-dioxygenase in Saccharomyces cerevisiae. Can. J. Microbiol. 1995, 41, 19–26. [Google Scholar] [CrossRef]
- Almeida, A.M.; Marchiosi, R.; Abrahão, J.; Constantin, R.P.; dos Santos, W.D.; Ferrarese-Filho, O. Revisiting the shikimate pathway and highlighting their enzyme inhibitors. Phytochem. Rev. 2023, 1–37. [Google Scholar] [CrossRef]
- Lynch, J.H.; Dudareva, N. Aromatic amino acids: A complex network ripe for future exploration. Trends Plant Sci. 2020, 25, 670–681. [Google Scholar] [CrossRef]
- Hubrich, F.; Müller, M.; Andexer, J.N. Chorismate- and isochorismate converting enzymes: Versatile catalysts acting on an important metabolic node. Chem. Commun. 2021, 57, 2441–2463. [Google Scholar] [CrossRef]
- Wu, S.; Chen, W.; Lu, S.; Zhang, H.; Yin, L. Metabolic engineering of shikimic acid biosynthesis pathway for the production of shikimic acid and its branched products in microorganisms: Advances and Prospects. Molecules 2022, 27, 4779. [Google Scholar] [CrossRef]
Figure 1.
The three NAD+ de novo biosynthesis pathways. Enzymes used in this work are indicated with blue. ADIC: 2-amino-2-deoxyisochorismate; DHHA: trans-2,3-dihydro-3-hydroxyanthranilic acid; 3-HAA: 3-hydroxyanthranilate; ACMS: 2-amino-3-carboxymuconate semialdehyde; PRPP: 5-phosphoribosyl diphosphate; NAMN: nicotinic acid mononucleotide; NAAD: nicotinic acid adenine dinucleotide.
Figure 1.
The three NAD+ de novo biosynthesis pathways. Enzymes used in this work are indicated with blue. ADIC: 2-amino-2-deoxyisochorismate; DHHA: trans-2,3-dihydro-3-hydroxyanthranilic acid; 3-HAA: 3-hydroxyanthranilate; ACMS: 2-amino-3-carboxymuconate semialdehyde; PRPP: 5-phosphoribosyl diphosphate; NAMN: nicotinic acid mononucleotide; NAAD: nicotinic acid adenine dinucleotide.
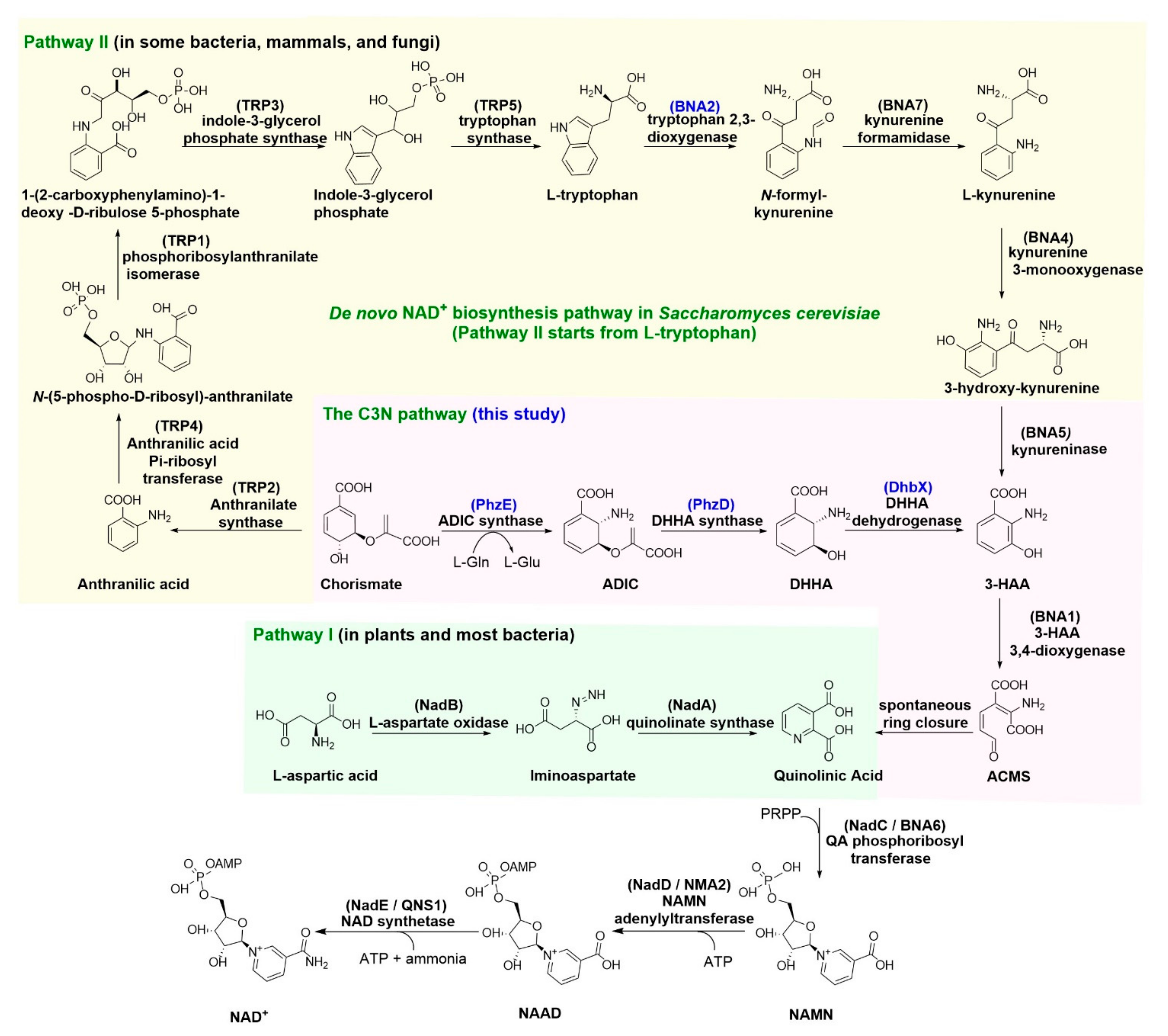
Figure 2.
Representative assay of the C3N pathway enzymes in S. cerevisiae. (A) Representative enzymatic assays using the crude proteins of wild type S. cerevisiae BY00-pYES2.0 (Con*) and BY00-phzE (PhzE*). (B) Representative enzymatic assays using the crude proteins of wild type S. cerevisiae BY00-pYES2.0 (Con*), BY00-phzD (PhzD*), and BY00-phzE (PhzE*). (C) Representative enzymatic assays using the proteins of DhbX*.
Figure 2.
Representative assay of the C3N pathway enzymes in S. cerevisiae. (A) Representative enzymatic assays using the crude proteins of wild type S. cerevisiae BY00-pYES2.0 (Con*) and BY00-phzE (PhzE*). (B) Representative enzymatic assays using the crude proteins of wild type S. cerevisiae BY00-pYES2.0 (Con*), BY00-phzD (PhzD*), and BY00-phzE (PhzE*). (C) Representative enzymatic assays using the proteins of DhbX*.
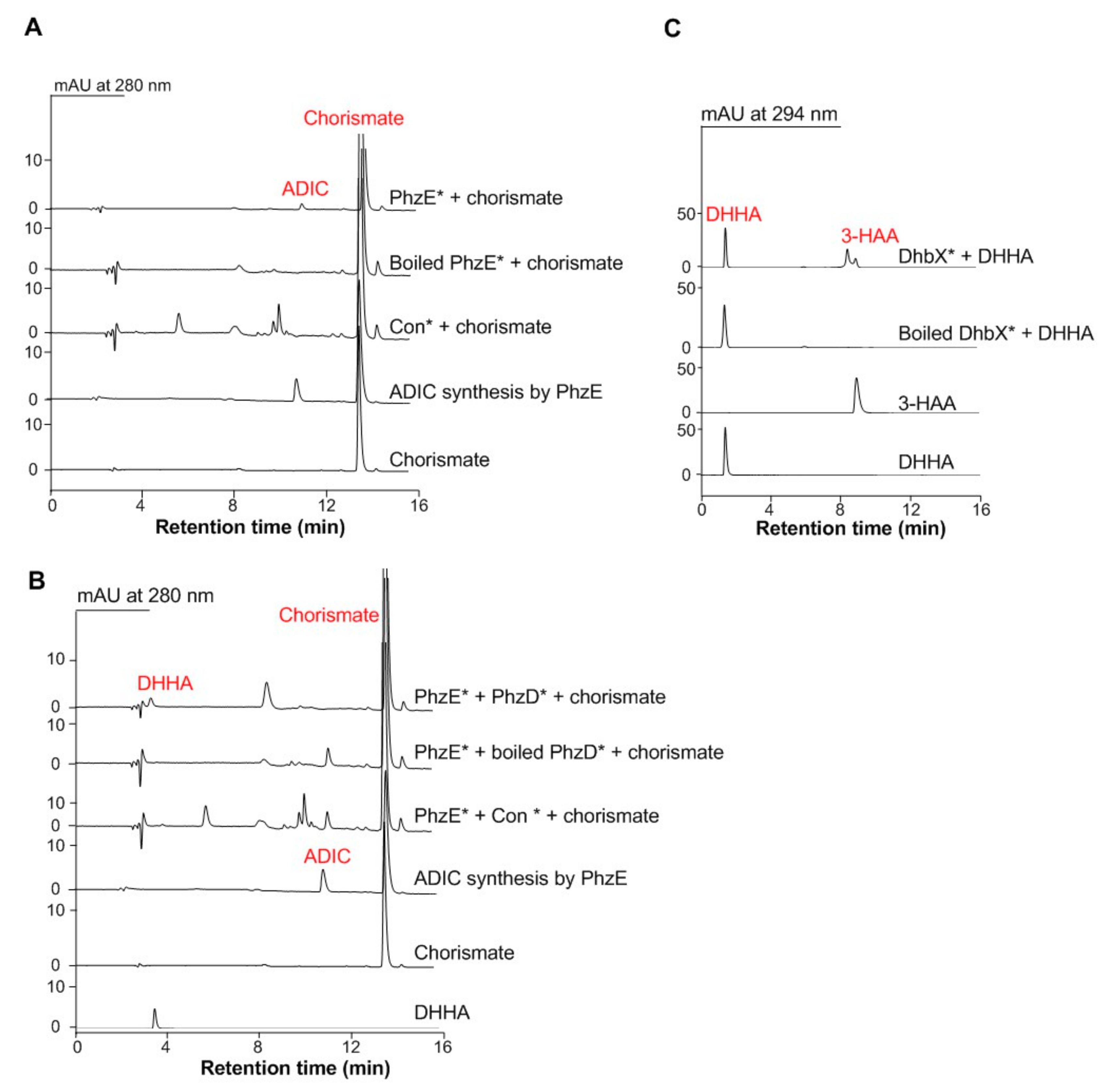
Figure 3.
Verification of the C3N pathway applicability in S. cerevisiae. (A) Genotype verification of the NAD+ auxotrophic S. cerevisiae strain BY01, in which the tryptophan 2,3-dioxygenase BNA2 was knocked out. Lane M, DNA marker; Lane 1 and lane 2 represent the PCR products of wild type S. cerevisiae BY00 and the ΔBNA2 mutant BY01, respectively. (B) Growth of S. cerevisiae BY00 (wild type), the ΔBNA2 mutant BY01, BY01-Con (control strain with empty vector), and BY01-C3N (with a complete C3N pathway) on YMMN plates with or without QA (10 mM) addition.
Figure 3.
Verification of the C3N pathway applicability in S. cerevisiae. (A) Genotype verification of the NAD+ auxotrophic S. cerevisiae strain BY01, in which the tryptophan 2,3-dioxygenase BNA2 was knocked out. Lane M, DNA marker; Lane 1 and lane 2 represent the PCR products of wild type S. cerevisiae BY00 and the ΔBNA2 mutant BY01, respectively. (B) Growth of S. cerevisiae BY00 (wild type), the ΔBNA2 mutant BY01, BY01-Con (control strain with empty vector), and BY01-C3N (with a complete C3N pathway) on YMMN plates with or without QA (10 mM) addition.
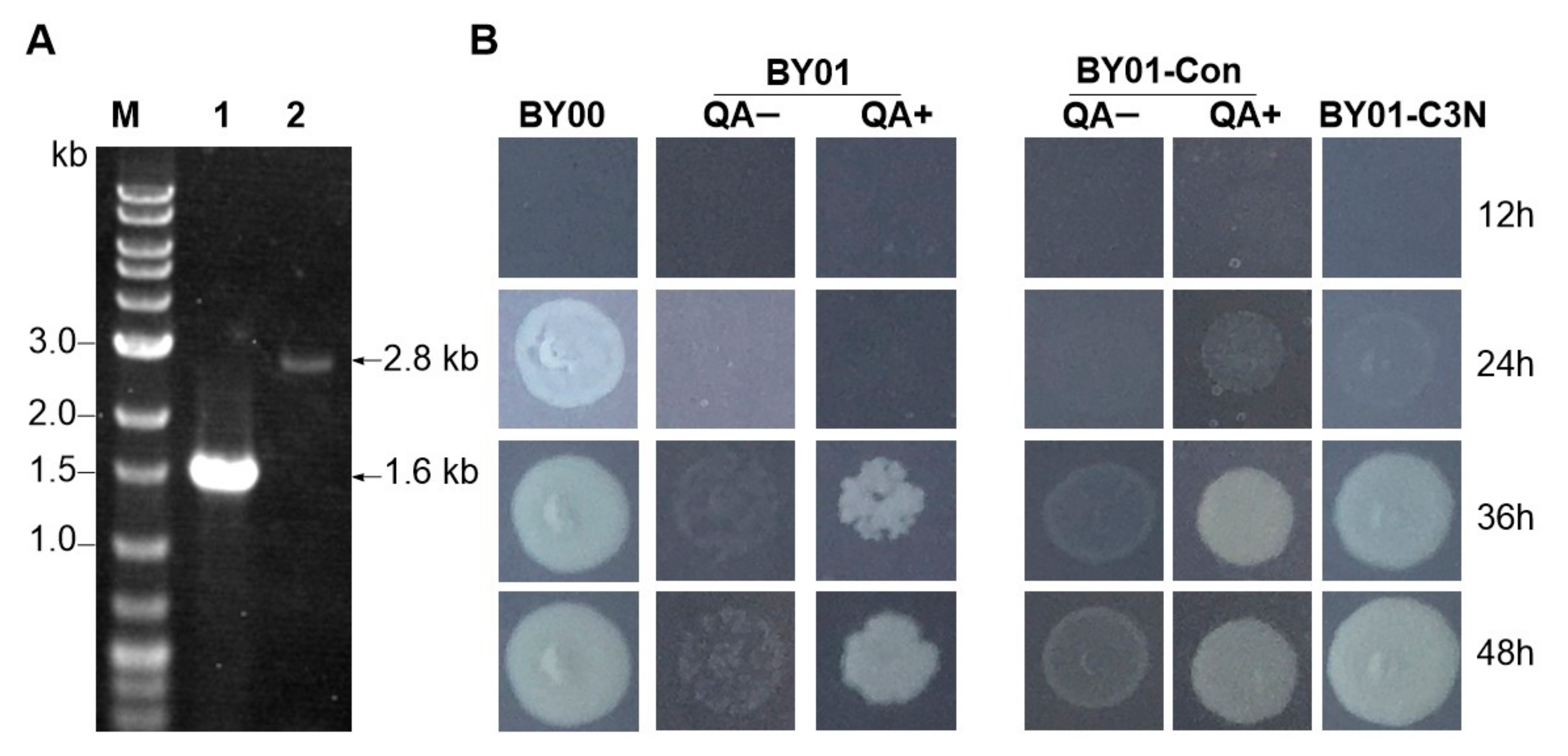
Figure 4.
Comparison of the growth curves and the cellular 3-HAA concentrations of different S. cerevisiae strains. (A) The growth curves of S. cerevisiae BY00 (wild type), BY00-Con (wild type with empty vector), BY01-C3N (ΔBNA2 mutant with a complete C3N pathway), and BY00-C3N (wild type with a complete C3N pathway) at 30 °C in YMMN medium with 20 g/L glucose addition. (B) The cellular 3-HAA concentrations of S. cerevisiae BY00-C3N and the control strains in 20 mL YMMN medium. Data presented as mean ± SD, n = 3, and p-values were calculated using two-tailed t-tests.
Figure 4.
Comparison of the growth curves and the cellular 3-HAA concentrations of different S. cerevisiae strains. (A) The growth curves of S. cerevisiae BY00 (wild type), BY00-Con (wild type with empty vector), BY01-C3N (ΔBNA2 mutant with a complete C3N pathway), and BY00-C3N (wild type with a complete C3N pathway) at 30 °C in YMMN medium with 20 g/L glucose addition. (B) The cellular 3-HAA concentrations of S. cerevisiae BY00-C3N and the control strains in 20 mL YMMN medium. Data presented as mean ± SD, n = 3, and p-values were calculated using two-tailed t-tests.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The cellular NAD(H) and NAD+ concentrations of different S. cerevisiae strains.
| Strains | NAD(H) [mM] | NAD+ [mM] |
|---|---|---|
| BY00 | 3.30 ± 0.45 | 1.90 ± 0.24 |
| BY00-Con | 2.92 ± 0.20 | 1.40 ± 0.19 |
| BY01-C3N | 3.13 ± 0.06 | 1.65 ± 0.09 |
| BY00-C3N | 4.59 ± 0.37 | 2.30 ± 0.25 |
Note: The standard deviation (mean ± SD) was calculated from independent triplicate biological experiments.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, X.; Tang, Y.; Ding, Y.; Li, P.; Chen, Y. Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae. Fermentation 2023, 9, 886. https://doi.org/10.3390/fermentation9100886
AMA Style
Li X, Tang Y, Ding Y, Li P, Chen Y. Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae. Fermentation. 2023; 9(10):886. https://doi.org/10.3390/fermentation9100886
Chicago/Turabian StyleLi, Xinli, Yue Tang, Yong Ding, Pengwei Li, and Yihua Chen. 2023. "Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae" Fermentation 9, no. 10: 886. https://doi.org/10.3390/fermentation9100886
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.