

Relationship between Structure and Zero-Field Splitting of Octahedral Nickel(II) Complexes with a Low-Symmetric Tetradentate Ligand

 , , , ,
, , , , 
Abstract
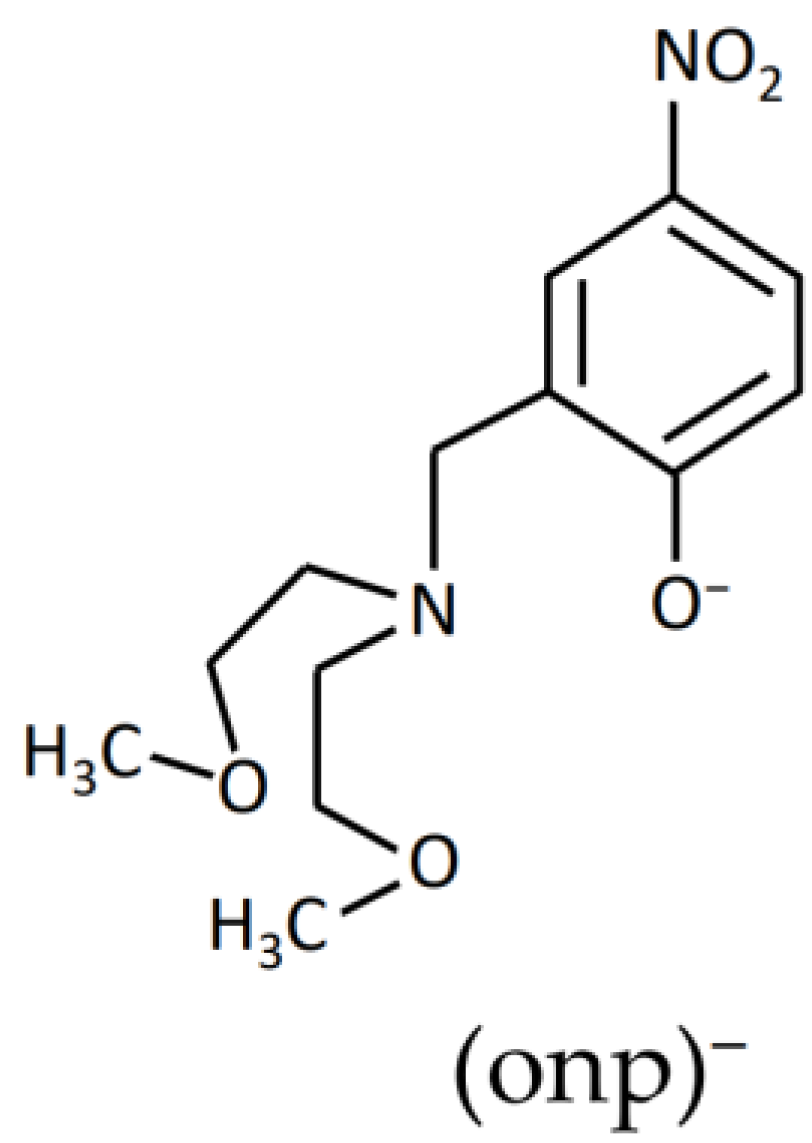
:1. Introduction
2. Materials and Methods
2.1. Measurements
2.2. Materials
2.3. Preparations
2.4. Crystallography
2.5. Computation
3. Results and Discussion
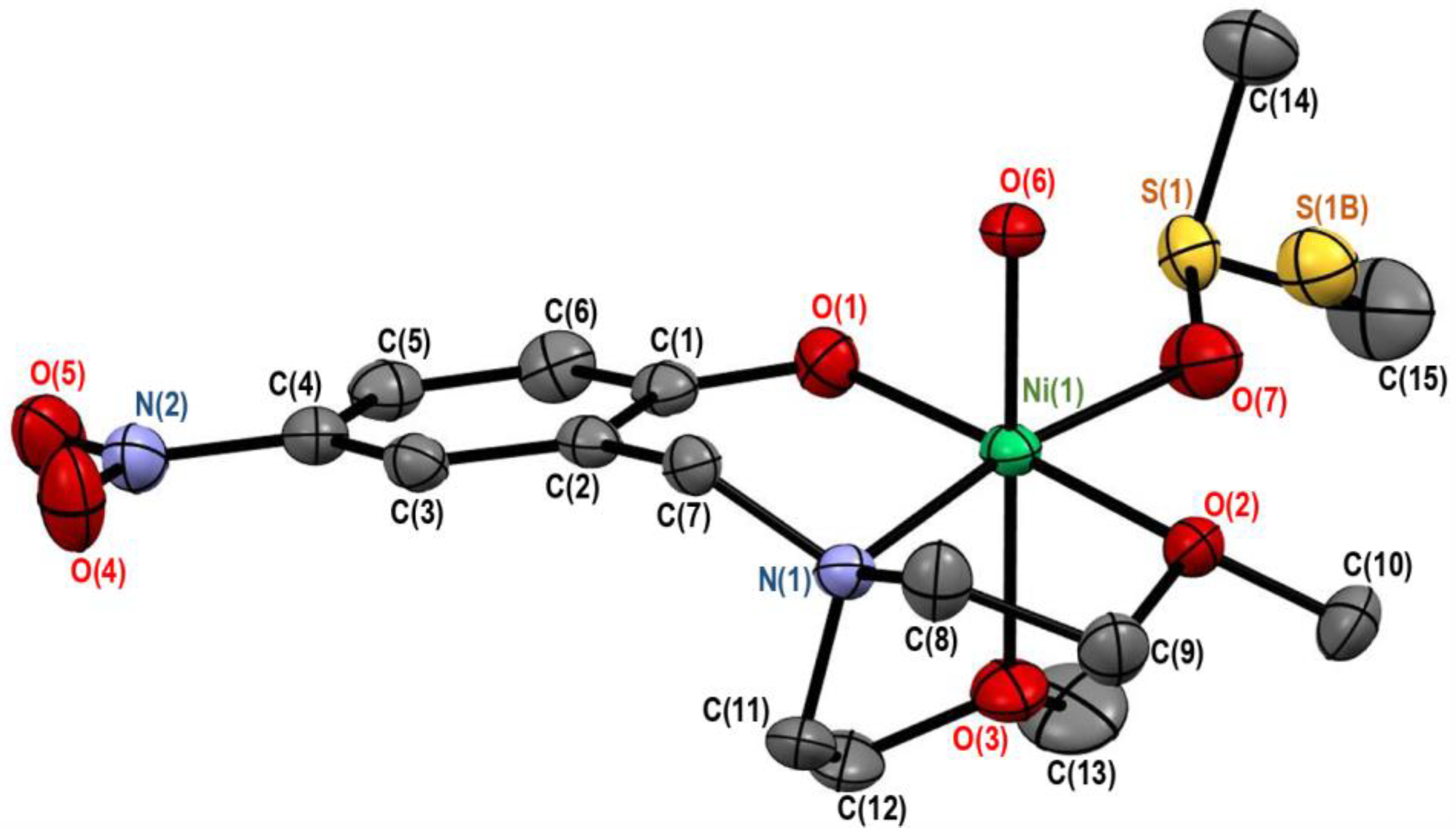
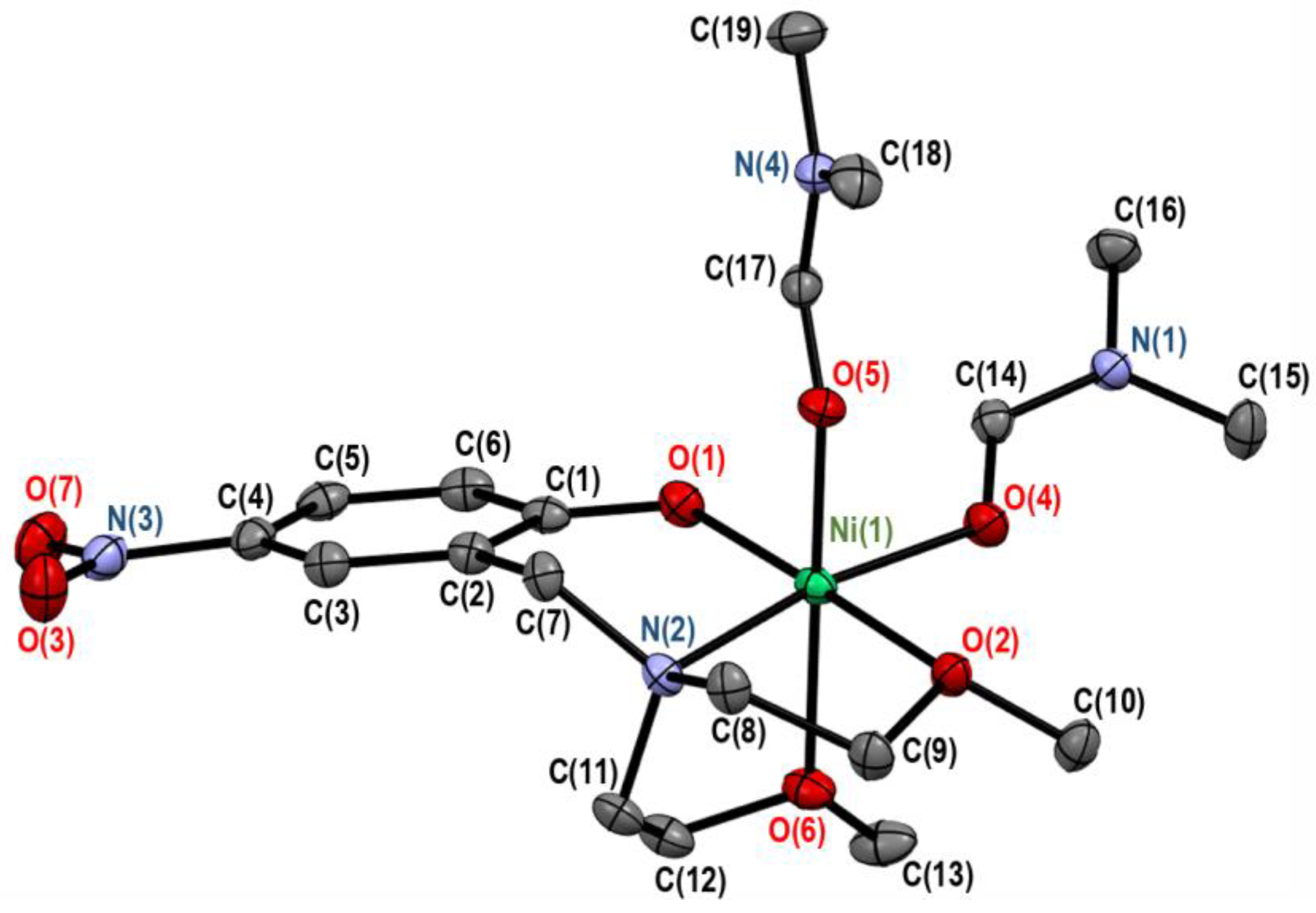
3.1. Crystal Structures of Complexes 2 and 3
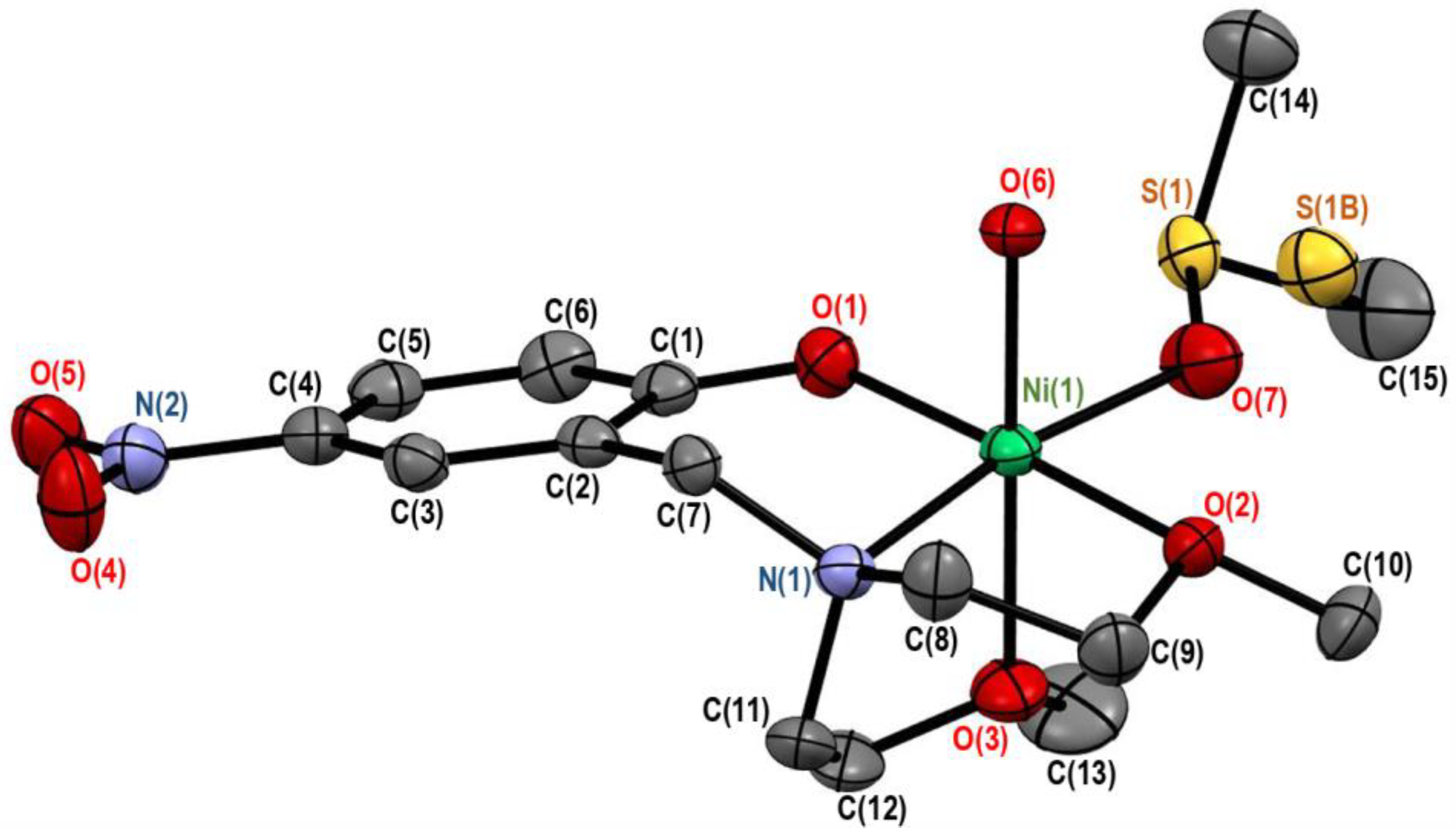
3.1.1. Crystal Structure of [Ni(onp)(dmso)(H2O)][BPh4]·dmso (2)
3.1.2. Crystal Structure of [Ni(onp)(dmf)2][BPh4] (3)
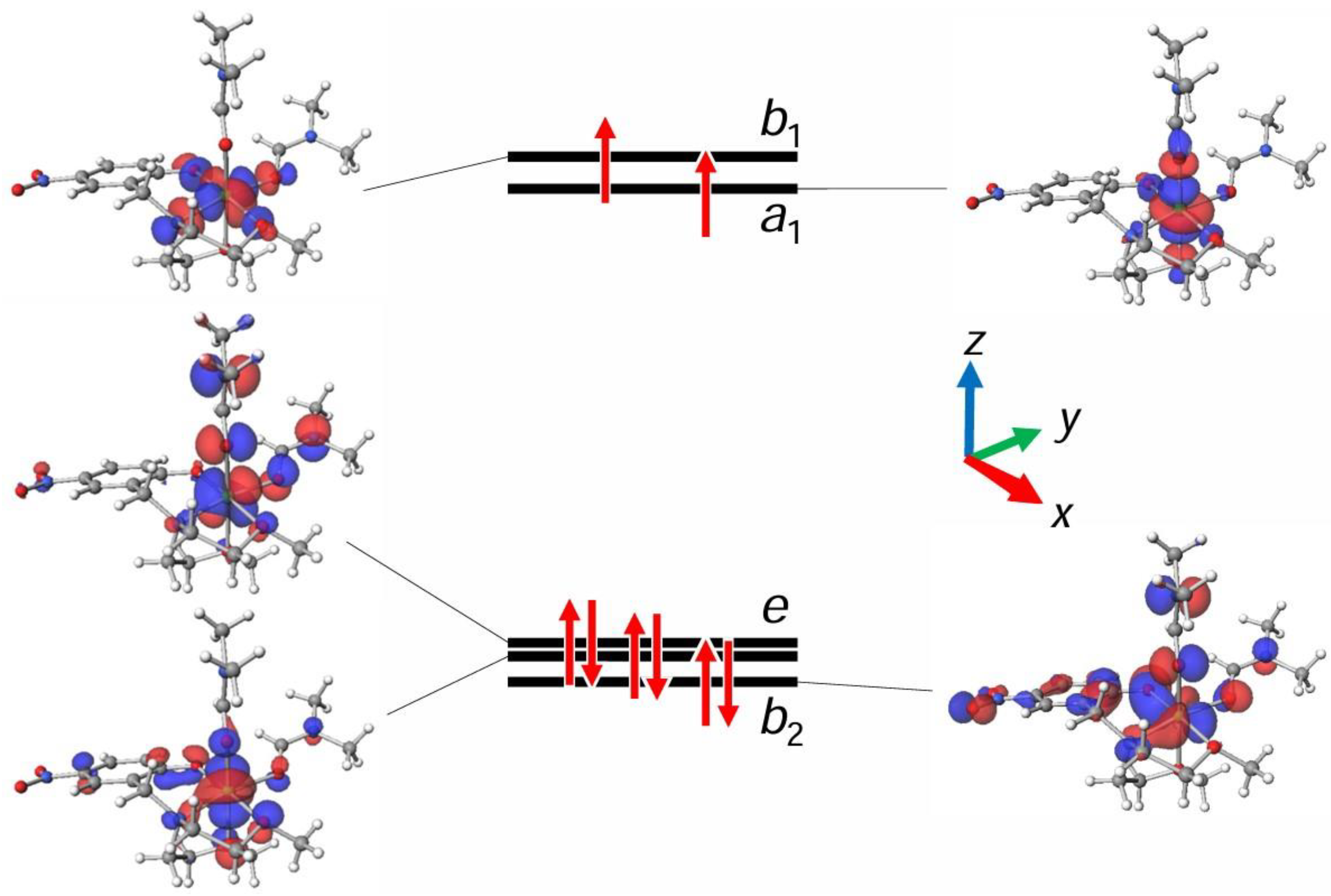
3.2. Density Functional Theory (DFT) Computations for Complexes 2 and 3
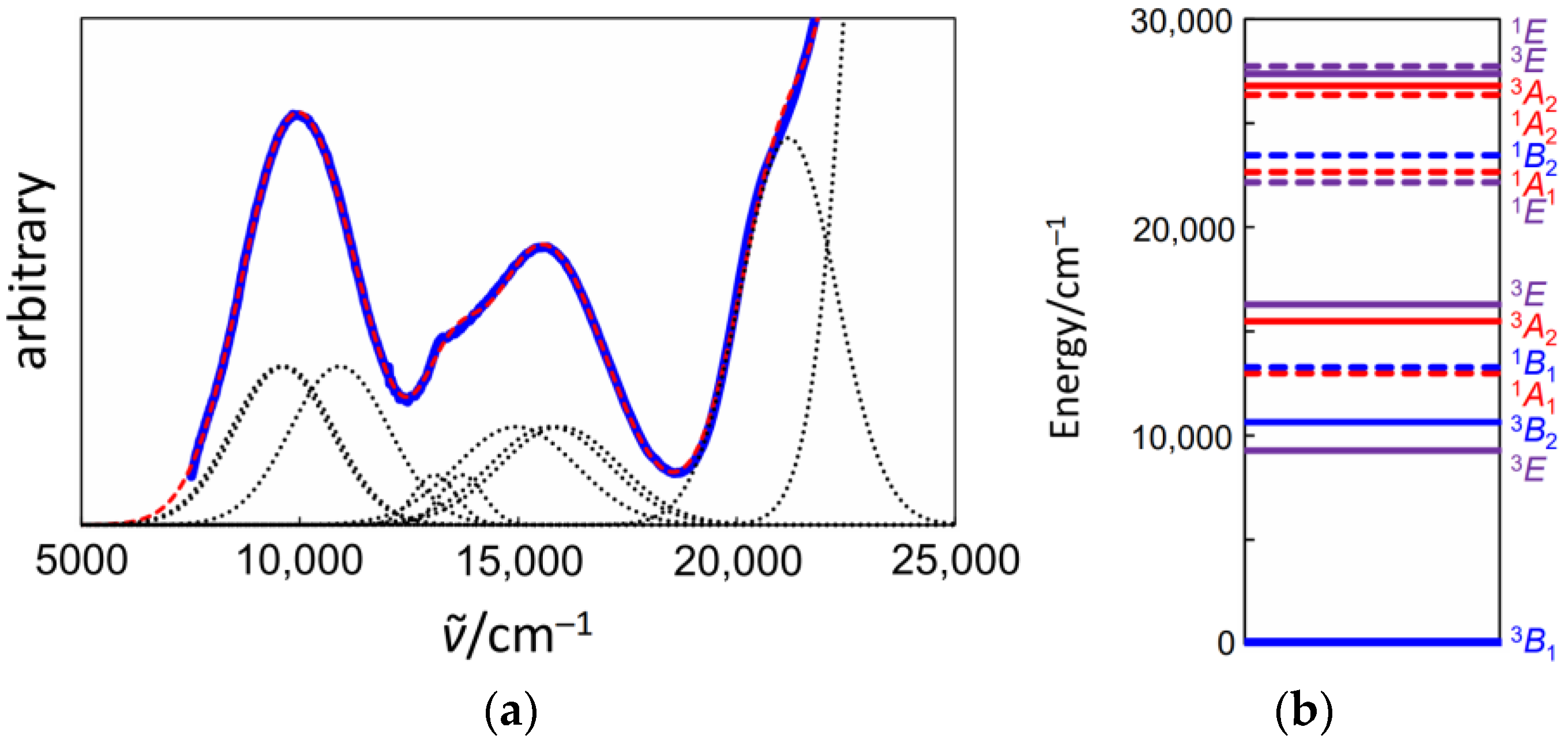
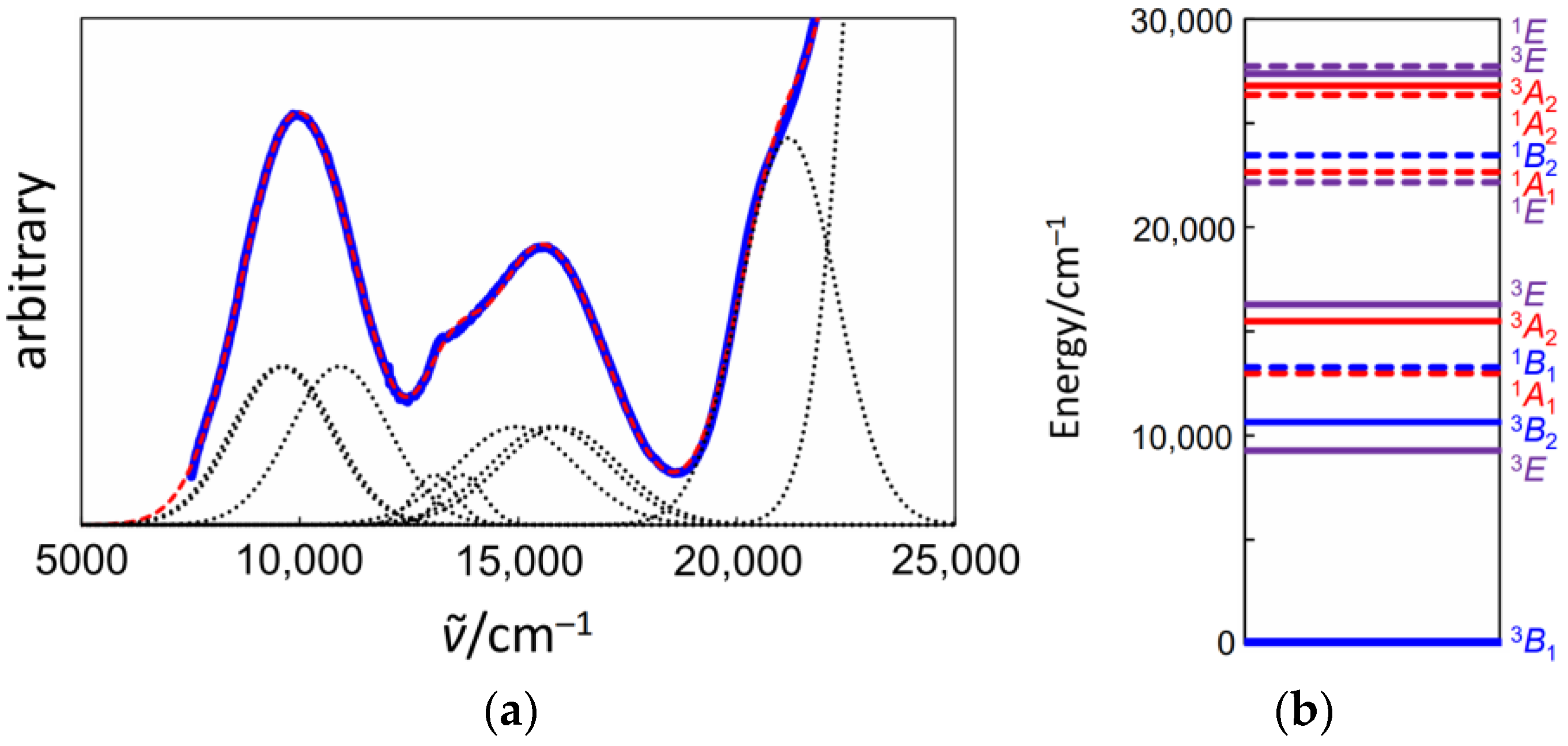
3.3. Electronic Spectra of 1
3.4. Theory and Magnetic Equations
| gz β Hu cosθ + D/3 | gx β Hu sinθ | 0 |
| gx β Hu sinθ | −2D/3 | gx β Hu sinθ |
| 0 | gx β Hu sinθ | −gz β Hu cosθ + D/3 |
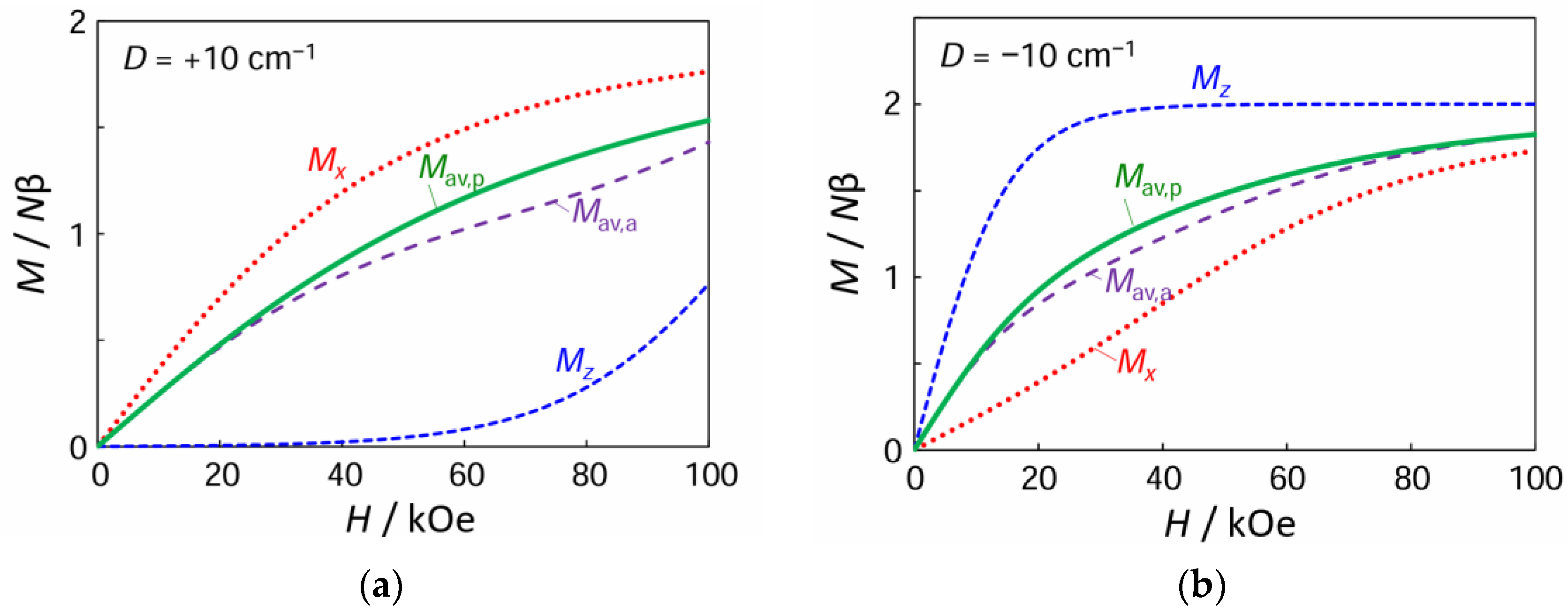
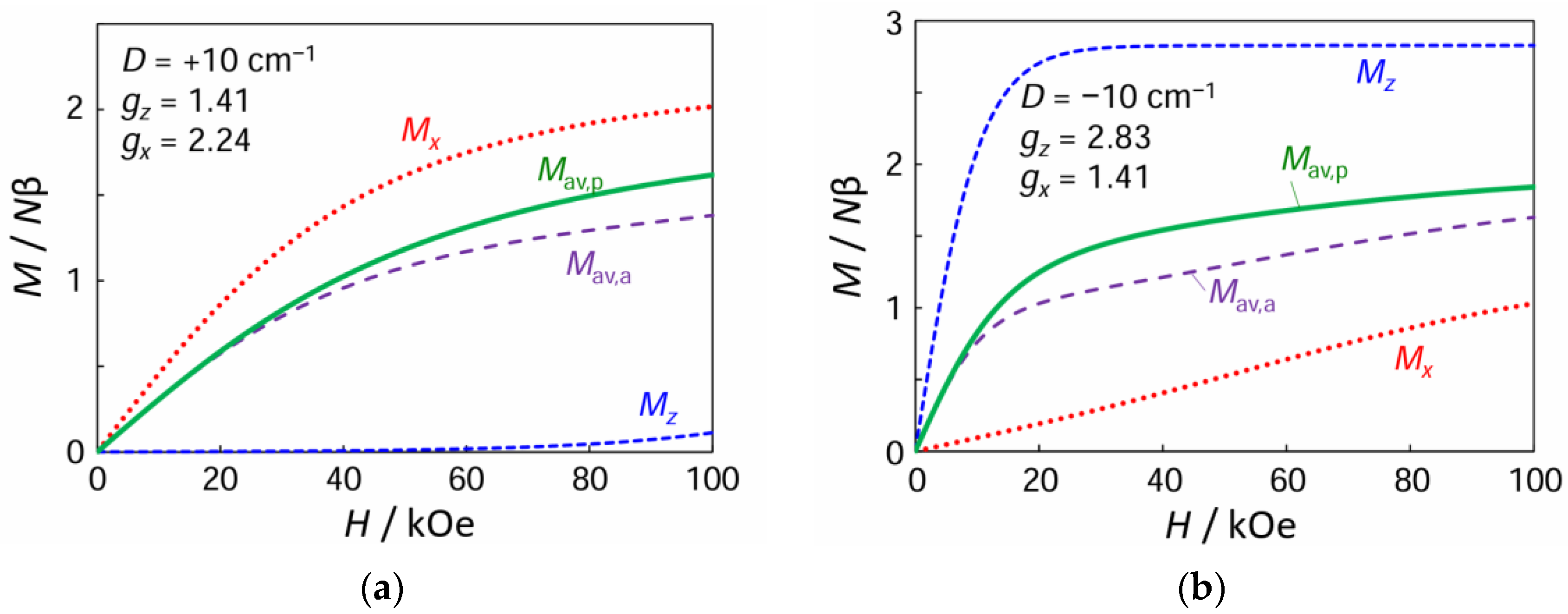
3.5. Magnetic Properties of Complex 1
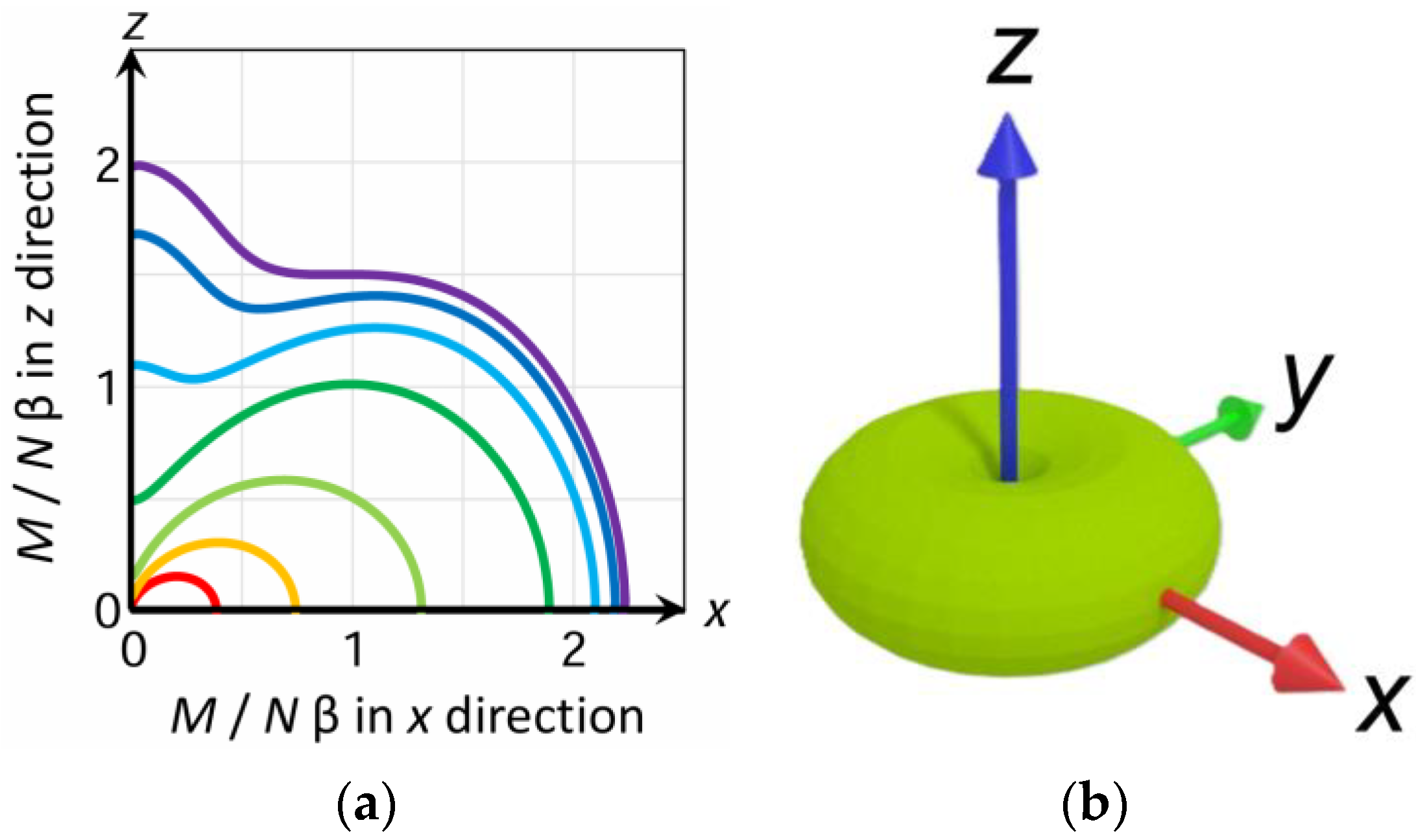
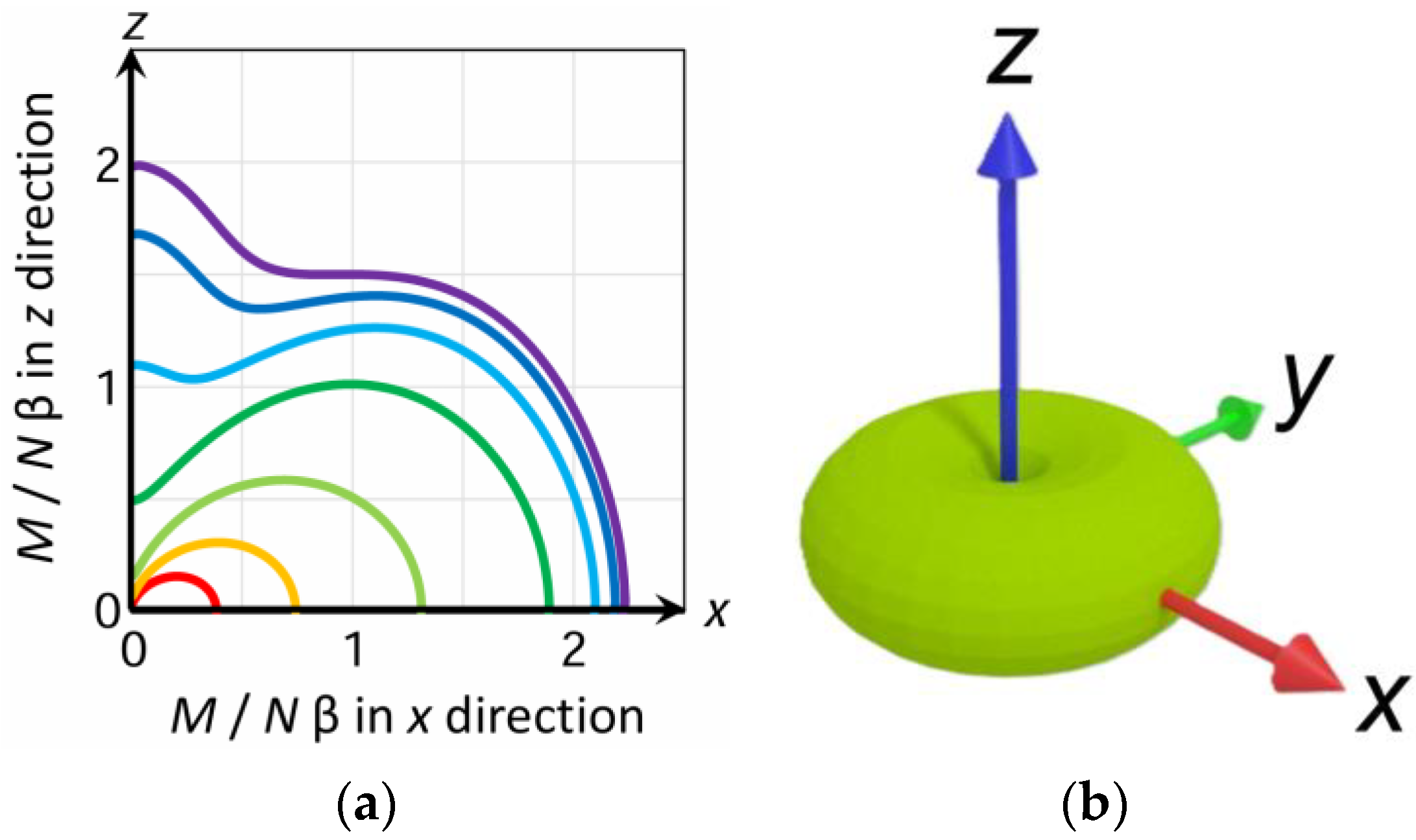
3.6. Powder Average Magnetization and Arithmetic Average Magnetization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
Appendix C
References
- Figgis, B.N.; Hitchman, M.A. Ligand Field Theory and Its Application; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar]
- Boča, R. Zero-field splitting in metal complexes. Coord. Chem. Rev. 2004, 248, 757–815. [Google Scholar] [CrossRef]
- Krzystek, J.; Ozarowski, A.; Telser, J. Multi-frequency, high-field EPR as a powerful tool to accurately determine zero-field splitting in high-spin transition metal coordination complexes. Coord. Chem. Rev. 2006, 250, 2308–2324. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; VCH Publishers, Inc.: New York, NY, USA, 1993; pp. 17–21. [Google Scholar]
- Sakiyama, H.; Chiba, Y.; Tone, K.; Yamasaki, M.; Mikuriya, M.; Krzystek, J.; Ozarowski, A. Magnetic Properties of a Dinuclear Nickel(II) Complex with 2,6-Bis[(2-hydroxyethyl)methylaminomethyl]-4-methylphenolate. Inorg. Chem. 2017, 56, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Coca, S.; Aravena, D.; Morales, R.; Ruiz, E.J. Large magnetic anisotropy in mononuclear metal complexes. Coord. Chem. Rev. 2015, 289–290, 379–392. [Google Scholar] [CrossRef]
- Zabala-Lekuona, A.; Seco, J.M.; Colacio, E. Single-Molecule Magnets: From Mn12-ac to dysprosium metallocenes, a travel in time. Coord. Chem. Rev. 2021, 441, 213984. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Sessoli, R.; Barra, A.L.; Brunel, L.C.; Guillot, M. Alternating current susceptibility, high field magnetization, and millimeter band EPR evidence for a ground S = 10 state in [Mn12O12(CH3COO)16(H2O)4]∙2CH3COOH∙4H2O. J. Am. Chem. Soc. 1991, 113, 5873–5874. [Google Scholar] [CrossRef]
- Sessoli, R.; Tsai, H.-L.; Schake, A.R.; Wang, S.; Vincent, J.B.; Folting, K.; Gatteschi, D.; Christou, G.; Hendrickson, D.N. High-Spin Molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 1993, 115, 1804–1816. [Google Scholar] [CrossRef]
- Boča, R.; Rajnák, C. Unexpected behavior of single ion magnets. Coord. Chem. Rev. 2021, 430, 213657. [Google Scholar] [CrossRef]
- Sakiyama, H.; Yamamoto, Y.; Hoshikawa, R.; Mitsuhashi, R. Crystal Structures and Magnetic Properties of Diaquatetrapyridinenickel(II) and Diaquatetrapyridinecobalt(II) Complexes. Magnetochemistry 2023, 9, 14. [Google Scholar] [CrossRef]
- Sanyal, R.; Kundu, P.; Rychagova, E.; Zhigulin, G.; Ketkov, S.; Ghosh, B.; Chattopadhyay, S.K.; Zangrando, E.; Das, D. Catecholase activity of Mannich-based dinuclear CuII complexes with theoretical modeling: New insight into the solvent role in the catalytic cycle. New J. Chem. 2016, 40, 6623–6635. [Google Scholar] [CrossRef]
- Sakiyama, H.; Mochizuki, R.; Sugawara, A.; Sakamoto, M.; Nishida, Y.; Yamasaki, M. Dinuclear zinc(II) complex of a new acyclic phenol-based dinucleating ligand with four methoxyethyl chelating arms: First dizinc model with aminopeptidase function. J. Chem. Soc. Dalton Trans. 1999, 997–1000. [Google Scholar] [CrossRef]
- Sakiyama, H.; Igarashi, Y.; Nakayama, Y.; Hossain, M.d.J.; Unoura, K.; Nishida, Y. Aminopeptidase function of dinuclear zinc(II) complexes of phenol-based dinucleating ligands: Effect of p-substituents. Inorg. Chim. Acta 2003, 351, 256–260. [Google Scholar] [CrossRef]
- Bain, G.A.; Berry, J.F. Diamagnetic corrections and Pascal’s constants. J. Chem. Educ. 2008, 85, 532. [Google Scholar] [CrossRef]
- Hoshikawa, R.; Mitsuhashi, R.; Asato, E.; Liu, J.; Sakiyama, H. Structures of dimer-of-dimers type defect cubane tetranuclear copper(II) complexes with novel dinucleating ligands. Molecules 2022, 27, 576. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in Electronic Structure Theory; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, H. Development of MagSaki software for magnetic analysis of dinuclear high-spin cobalt(II) complexes in an axially distorted octahedral field. J. Chem. Software 2001, 7, 171–178. [Google Scholar] [CrossRef]
- Sakiyama, H. Development of MagSaki(A) software for the magnetic analysis of dinuclear high-spin cobalt(II) complexes considering anisotropy in exchange interaction. J. Comput. Chem. Jpn. 2007, 6, 123–134. [Google Scholar] [CrossRef]
- Sakiyama, H. Development of MagSaki(Tri) software for the magnetic analysis of trinuclear high-spin cobalt(II) complexes. J. Comput. Chem. Jpn. Int. Ed. 2015, 1, 9–13. [Google Scholar] [CrossRef]
- Sakiyama, H. Development of MagSaki(Tetra) software for the magnetic analysis of tetranuclear high-spin cobalt(II) complexes. J. Comput. Chem. Jpn. Int. Ed. 2016, 2, 2016-0001. [Google Scholar] [CrossRef]
- Sakiyama, H.; Sudo, R.; Abiko, T.; Yoshioka, D.; Mitsuhashi, R.; Omote, M.; Mikuriya, M.; Yoshitake, M.; Koikawa, M. Magneto-structural correlation of hexakis-dmso cobalt(II) complex. Dalton Trans. 2017, 46, 16306. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, H.; Abiko, T.; Ito, M.; Mitsuhashi, R.; Mikuriya, M.; Waki, K.; Usuki, T. Reversible crystal-to-crystal phase transition of an octahedral zinc(II) complex with six dimethylsulfoxide. Polyhedron 2019, 158, 494–498. [Google Scholar] [CrossRef]
- Sakiyama, H.; Suzuki, T.; Ono, K.; Ito, R.; Watanabe, Y.; Yamasaki, M.; Mikuriya, M. Synthesis, structure, and magnetic properties of dinuclear nickel(II) complexes with a phenol-based dinucleating ligand with four methoxyethyl chelating arms. Inorg. Chim. Acta 2005, 358, 1897–1903. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Tasaki, M.; Asato, E.; Sakiyama, H. Synthesis and Crystal Structure of a Nickel(II) Complex with Bis(2-methoxyethyl)amine. X-ray Struct. Anal. Online 2011, 27, 5–6. [Google Scholar] [CrossRef]
- Bendix, J.; Schäffer, C.E.; Brorson, M. Quantitative formulation of ligand field theory by the use of orthonormal operators. Exemplification by means of pq systems. Coord. Chem. Rev. 1989, 94, 181–241. [Google Scholar] [CrossRef]
- Kudrat-E-Zahan, M.; Nishida, Y.; Sakiyama, H. Identification of cis/trans isomers of bis(acetylacetonato)nickel(II) complexes in solution based on electronic spectra. Inorg. Chim. Acta 2010, 363, 168–172. [Google Scholar] [CrossRef]
- Mabbs, F.E.; Machin, D.J. Magnetism and Transition Metal Complexes; Dover Publications, Inc.: Mineola, NY, USA, 2008. [Google Scholar]
- Griffith, J.S. The Theory of Transition-Metal Ions; Cambridge University Press: Cambridge, UK, 1961. [Google Scholar]
- Sakiyama, H.; Abiko, T.; Yoshida, K.; Shomura, K.; Mitsuhashi, R.; Koyama, Y.; Mikuriya, M.; Koikawa, M.; Mitsumi, M. Detailed magnetic analysis and successful deep-neural-network-based conformational prediction for [VO(dmso)5][BPh4]2. RSC Adv. 2020, 10, 9678–9685. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Complex 2 | Complex 3 |
|---|---|---|
| Empirical Formula | C41H53BN2NiO8S2 | C43H53BN4NiO7 |
| Formula Weight | 835.49 | 807.41 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21/n | P21/c |
| a/Å | 11.5855 (3) | 12.6151 (2) |
| b/Å | 16.3220 (4) | 16.2543 (2) |
| c/Å | 22.1213 (5) | 20.7114 (3) |
| β/° | 99.695 (2) | 106.906 (2) |
| V/Å3 | 4123.36 (18) | 4063.33 (11) |
| Z | 4 | 4 |
| Crystal Dimensions/mm | 0.164 × 0.114 × 0.057 | 0.138 × 0.098 × 0.064 |
| T/K | 100 | 100 |
| λ/Å | 0.71073 | 0.71073 |
| ρcalcd/g cm−3 | 1.346 | 1.320 |
| µ/mm−1 | 0.625 | 0.533 |
| F(000) | 1768 | 1712 |
| 2θmax/◦ | 55 | 60 |
| No. of Reflections Measured | 28,549 | 60,987 |
| No. of independent reflections | 9416 (Rint = 0.0298) | 10,822 (Rint = 0.0320) |
| Data/restraints/parameters | 9416/3/518 | 10,822/0/511 |
| R1 (I > 2.00σ(I)) 1 | 0.0409 | 0.0297 |
| wR2 (All reflections) 2 | 0.1093 | 0.0852 |
| Goodness of Fit Indicator | 1.045 | 1.044 |
| Highest peak, deepest hole/e Å−3 | 1.001, −0.539 | 0.413, −0.416 |
| CCDC deposition number | 2335894 | 2335893 |
| Atom–Atom | Distance/Å | Atom–Atom | Distance/Å |
|---|---|---|---|
| Ni(1)–O(1) | 1.9957(14) | Ni(1)–O(2) | 2.1199 (14) |
| Ni(1)–O(3) | 2.1121(15) | Ni(1)–O(6) | 2.0708 (14) |
| Ni(1)–O(7) | 2.0261(16) | Ni(1)–N(1) | 2.0583 (17) |
| Atom–Atom–Atom | Angle/° | Atom–Atom–Atom | Angle/° |
|---|---|---|---|
| O(1)–Ni(1)–O(2) | 176.09 (6) | O(1)–Ni(1)–O(3) | 93.37 (6) |
| O(1)–Ni(1)–O(6) | 91.60 (6) | O(1)–Ni(1)–O(7) | 95.30 (7) |
| O(1)–Ni(1)–N(1) | 93.15 (6) | O(2)–Ni(1)–O(3) | 87.02 (6) |
| O(2)–Ni(1)–O(6) | 87.68 (6) | O(2)–Ni(1)–O(7) | 88.54 (7) |
| O(2)–Ni(1)–N(1) | 83.07 (6) | O(3)–Ni(1)–O(6) | 172.88 (6) |
| O(3)–Ni(1)–O(7) | 94.28 (7) | O(3)–Ni(1)–N(1) | 80.59 (6) |
| O(6)–Ni(1)–O(7) | 90.33 (6) | O(6)–Ni(1)–N(1) | 94.06 (6) |
| O(7)–Ni(1)–N(1) | 170.36 (7) |
| Atom–Atom | Distance/Å | Atom–Atom | Distance/Å |
|---|---|---|---|
| Ni(1)–O(1) | 1.9906 (8) | Ni(1)–O(2) | 2.1330 (8) |
| Ni(1)–O(4) | 2.0295 (8) | Ni(1)–O(5) | 2.0709 (8) |
| Ni(1)–O(6) | 2.1240 (8) | Ni(1)–N(2) | 2.0643 (9) |
| Atom–Atom–Atom | Angle/° | Atom–Atom–Atom | Angle/° |
|---|---|---|---|
| O(1)–Ni(1)–O(2) | 177.10 (3) | O(1)–Ni(1)–O(4) | 93.97 (3) |
| O(1)–Ni(1)–O(5) | 93.28 (3) | O(1)–Ni(1)–O(6) | 94.85 (3) |
| O(1)–Ni(1)–N(2) | 94.22 (3) | O(2)–Ni(1)–O(4) | 88.85 (3) |
| O(2)–Ni(1)–O(5) | 86.00 (3) | O(2)–Ni(1)–O(6) | 85.53 (3) |
| O(2)–Ni(1)–N(2) | 83.01(3) | O(4)–Ni(1)–O(5) | 91.61 (3) |
| O(4)–Ni(1)–O(6) | 94.96 (3) | O(4)–Ni(1)–N(2) | 170.58 (3) |
| O(5)–Ni(1)–O(6) | 169.17 (3) | O(5)–Ni(1)–N(2) | 92.55 (3) |
| O(6)–Ni(1)–N(2) | 79.72 (3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakiyama, H.; Kimura, R.; Oomiya, H.; Mitsuhashi, R.; Fujii, S.; Kanaizuka, K.; Muddassir, M.; Tamaki, Y.; Asato, E.; Handa, M. Relationship between Structure and Zero-Field Splitting of Octahedral Nickel(II) Complexes with a Low-Symmetric Tetradentate Ligand. Magnetochemistry 2024, 10, 32. https://doi.org/10.3390/magnetochemistry10050032
Sakiyama H, Kimura R, Oomiya H, Mitsuhashi R, Fujii S, Kanaizuka K, Muddassir M, Tamaki Y, Asato E, Handa M. Relationship between Structure and Zero-Field Splitting of Octahedral Nickel(II) Complexes with a Low-Symmetric Tetradentate Ligand. Magnetochemistry. 2024; 10(5):32. https://doi.org/10.3390/magnetochemistry10050032
Chicago/Turabian StyleSakiyama, Hiroshi, Rin Kimura, Haruto Oomiya, Ryoji Mitsuhashi, Sho Fujii, Katsuhiko Kanaizuka, Mohd. Muddassir, Yuga Tamaki, Eiji Asato, and Makoto Handa. 2024. "Relationship between Structure and Zero-Field Splitting of Octahedral Nickel(II) Complexes with a Low-Symmetric Tetradentate Ligand" Magnetochemistry 10, no. 5: 32. https://doi.org/10.3390/magnetochemistry10050032