
First-Principles Point Defect Models for Zr7Ni10 and Zr2Ni7 Phases



Abstract
:
1. Introduction
2. Computational Details
3. Experimental Setup
4. Results and Discussion
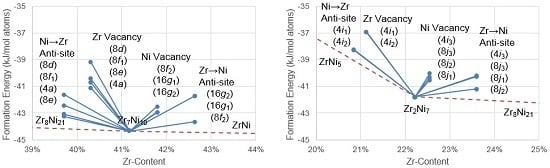
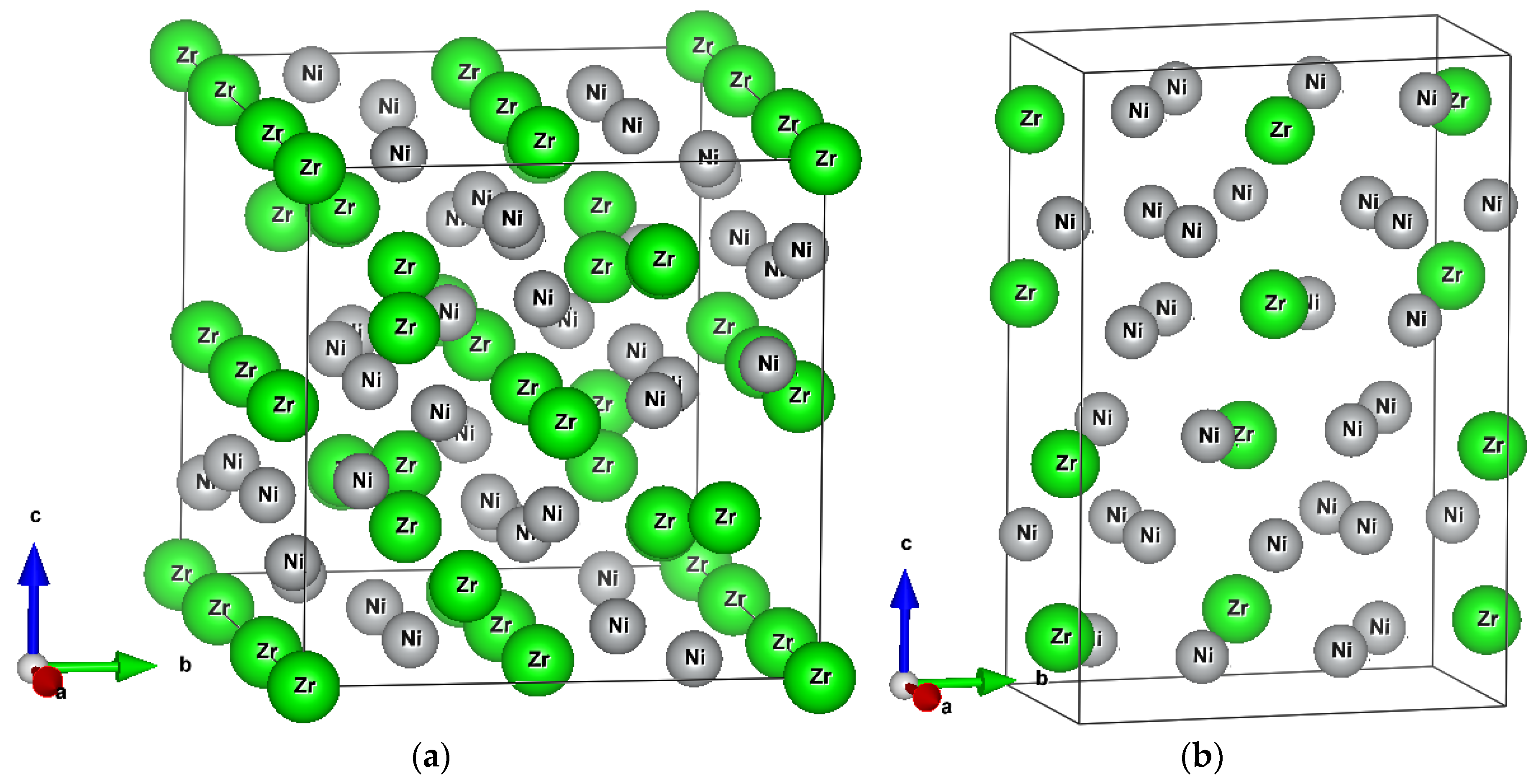
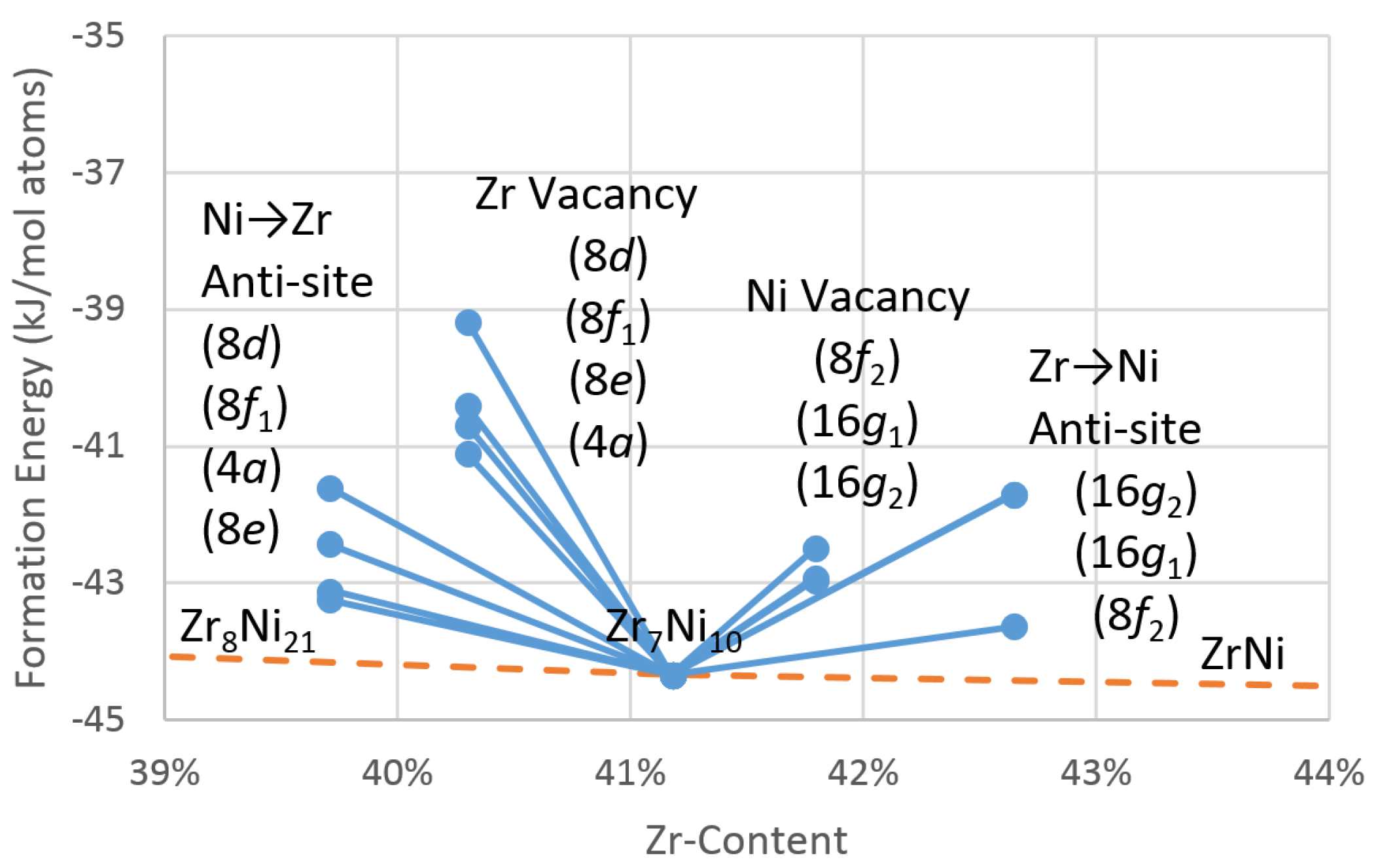
4.1. Theoretical Point Defects in Zr7Ni10
4.1.1. Density Functional Theory Defect Formation Energies
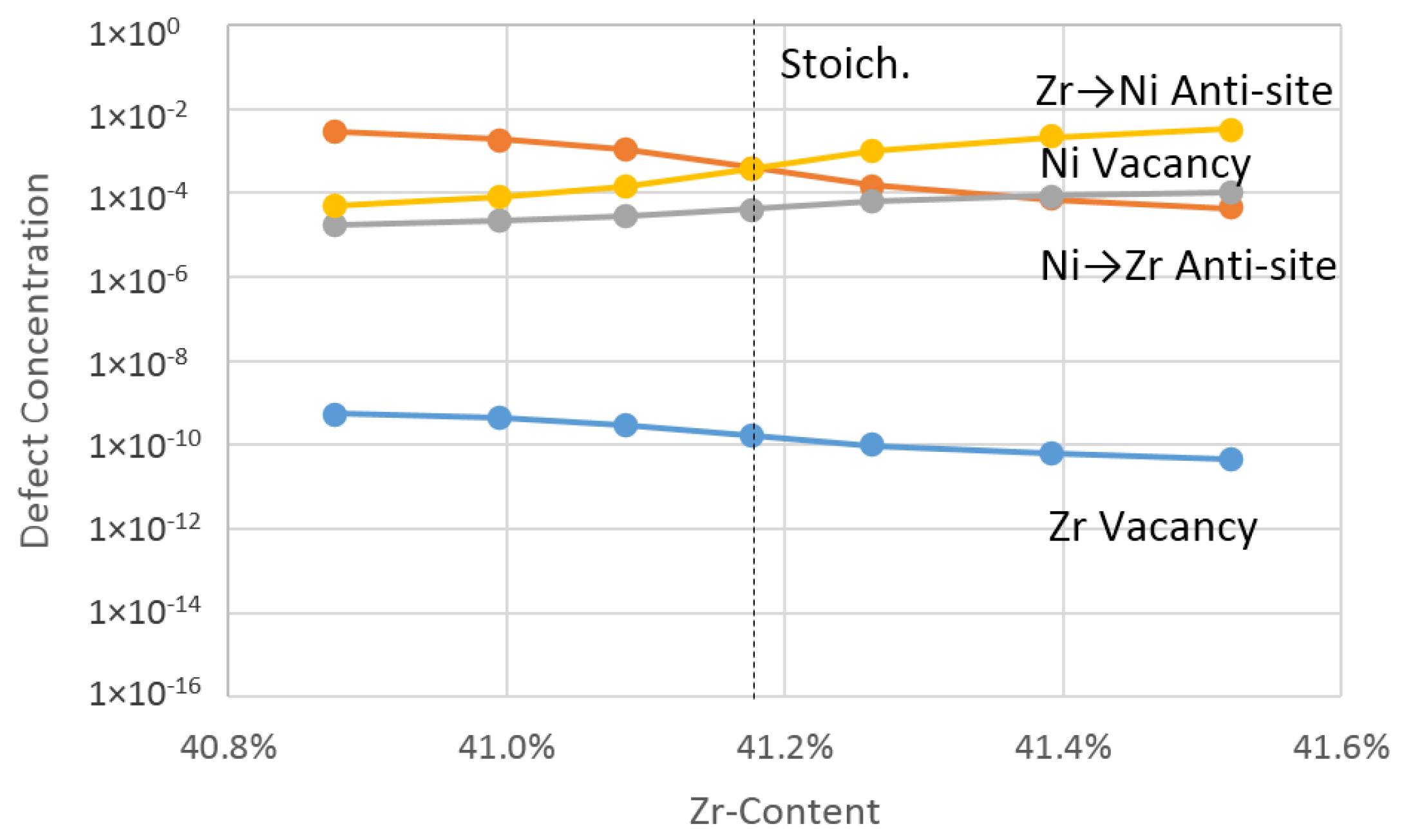
4.1.2. Theoretical Effective Defect Formation Model
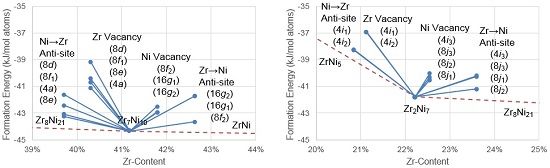
4.2. Theoretical Point Defects in Zr2Ni7
4.2.1. Density Functional Theory Formation Energies
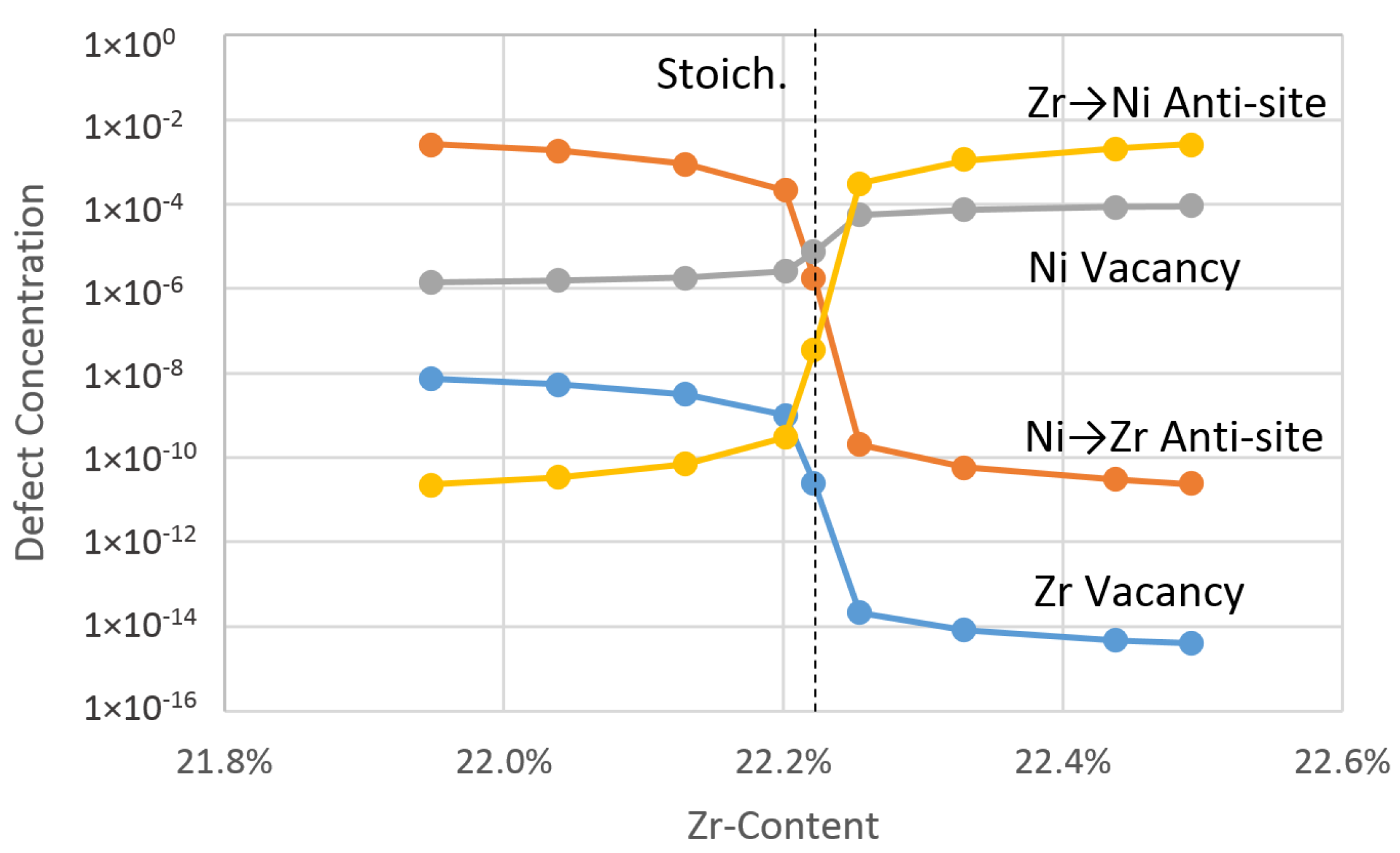
4.2.2. Theoretical Effective Defect Formation Model
4.3. Experimental Zr-Rich and Ni-Rich Zr7Ni10 Phase Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix
Abbreviations
| Concentration of defect i on the sublattice ν | |
| Concentration of vacancies on the Ni sublattices | |
| Concentration of vacancies on the Zr sublattices | |
| Concentration of Zr anti-site atoms in the Ni sublattices | |
| Concentration of Ni anti-site atoms in the Zr sublattices | |
| DFT | Density functional theory |
| EDS | Energy dispersive spectroscopy |
| J’ | Generalized grand canonical potential |
| kB | Boltzmann constant |
| M | Number of lattice sites |
| NA | Number of A atoms |
| NB | Number of B atoms |
| Number of defect i on the sublattice ν | |
| MH | Metal hydride |
| Ni/MH | Nickel/metal hydride |
| P | Pressure |
| Entropy per atom for non-defected structure | |
| Defect entropy formation parameter for defect i on sublattice ν | |
| SEM | Scanning electron microscopy |
| T | Temperature |
| U | Internal energy |
| Unit-cell volume per atom for non-defected structure | |
| Defect volume formation parameter for defect i on sublattice ν | |
| XRD | X-ray diffraction |
| Z | Number of formula units |
| αk | kth sublattice for A-site atoms |
| βl | lth sublattice for B-site atoms |
| ΔHf | Energy of formation |
| ΔHvNi | Effective defect formation energy for vacancies in the Ni sublattices |
| ΔHvZr | Effective defect formation energy for vacancies in the Zr sublattices |
| ΔHZrNi | Effective defect formation energy for Zr anti-site atoms in the Ni sublattices |
| ΔHNiZr | Effective defect formation energy for Ni anti-site atoms in the Zr sublattices |
| Energy per atom for non-defected structure | |
| Defect energy formation parameter for defect i on sublattice ν | |
| µA | Chemical potential for A atom |
| µB | Chemical potential for B atom |
References
- Joubert, J.M.; Latroche, M.; Percheron-Guégan, A.; Bouet, J. Improvement of the electrochemical activity of Zr–Ni–Cr laves phase hydride electrodes by secondary phase precipitation. J. Alloy. Compd. 1996, 240, 219–228. [Google Scholar] [CrossRef]
- Young, K.; Nei, J.; Ouchi, T.; Fetcenko, M. Phase abundances in AB2 metal hydride alloys and their correlations to various properties. J. Alloy. Compd. 2011, 509, 2277–2284. [Google Scholar] [CrossRef]
- Young, K.; Ouchi, T.; Fetcenko, M.A.; Mays, W.; Reichman, B. Structural and electrochemical properties of Ti1.5Zr5.5VxNi10−x. Int. J. Hydrog. Energy 2009, 34, 8695–8706. [Google Scholar] [CrossRef]
- Joubert, J.M.; Latroche, M.; Percheron-Guégan, A. Hydrogen absorption properties of several intermetallic compounds of the Zr Ni system. J. Alloy. Compd. 1995, 231, 494–497. [Google Scholar] [CrossRef]
- Young, K.; Ouchi, T.; Liu, Y.; Reichman, B.; Mays, W.; Fetcenko, M. Structural and electrochemical properties of TixZr7−xNi10. J. Alloy. Compd. 2009, 480, 521–528. [Google Scholar] [CrossRef]
- Young, M.; Chang, S.; Young, K.; Nei, J. Hydrogen storage properties of ZrVxNi3.5−x (x = 0.0–0.9) metal hydride alloys. J. Alloy. Compd. 2013, 580, S171–S174. [Google Scholar] [CrossRef]
- Myers, S.M.; Baskes, M.I.; Birnbaum, H.K.; Corbett, J.W.; DeLeo, G.G.; Estreicher, S.K.; Haller, E.E.; Jena, P.; Johnson, N.M.; Kirchheim, R.; et al. Hydrogen interactions with defects in crystalline solids. Rev. Mod. Phys. 1992, 64, 559–617. [Google Scholar] [CrossRef]
- Chen, K.C.; Peterson, E.J.; Thoma, D.J. HfCo2 laves phase intermetallics—Part I: Solubility limits and defect mechanisms. Intermetallics 2001, 9, 771–783. [Google Scholar] [CrossRef]
- Chen, K.C.; Chu, F.; Kotula, P.G.; Thoma, D. HfCo2 laves phase intermetallics—Part II: Elastic and mechanical properties as a function of composition. Intermetallics 2001, 9, 785–798. [Google Scholar] [CrossRef]
- Ovshinsky, S.R.; Fetcenko, M.A.; Ross, J. A nickel metal hydride battery for electric vehicles. Science 1993, 260, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Züttel, A. Materials for hydrogen storage. Mater. Today 2003, 6, 24–33. [Google Scholar] [CrossRef]
- Young, K. Stoichiometry in inter-Metallic Compounds for Hydrogen Storage Applications. In Stoichiometry and Materials Science—When Numbers Matter; Innocenti, A., Kamarulzaman, N., Eds.; InTech: Rijeka, Croatia, 2012; pp. 147–172. [Google Scholar]
- Stein, F.; Palm, M.; Sauthoff, G. Structure and stability of Laves phases. Part I. Critical assessment of factors controlling Laves phase stability. Intermetallics 2004, 12, 713–720. [Google Scholar] [CrossRef]
- Moura, C.S.; Motta, A.T.; Lam, N.Q.; Amaral, L. Atomistic simulations of point defects in zrni intermetallic compounds. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2001, 180, 257–264. [Google Scholar] [CrossRef]
- Mayer, J.; Elsasser, C.; Fahnle, M. Concentrations of atomic defects in B2-FexAl1−x—An ab-initio study. Phys. Status Solidi B Basic Res. 1995, 191, 283–298. [Google Scholar] [CrossRef]
- Mayer, J.; Fahnle, M. On the meaning of effective formation energies, entropies and volumes for atomic defects in ordered compounds. Acta Mater. 1997, 45, 2207–2211. [Google Scholar] [CrossRef]
- Meyer, B.; Fähnle, M. Atomic defects in the ordered compound B2-NiAl: A combination of ab initio electron theory and statistical mechanics. Phys. Rev. B 1999, 59, 6072–6082. [Google Scholar] [CrossRef]
- Rasamny, M.; Weinert, M.; Fernando, G.; Watson, R. Electronic structure and thermodynamics of defects in NiAl3. Phys. Rev. B 2001, 64. [Google Scholar] [CrossRef]
- Notten, P.; Latroche, M.; Percheron-Guégan, A. The influence of Mn on the crystallography and electrochemistry of nonstoichiometric AB5-type hydride-forming compounds. J. Electrochem. Soc. 1999, 146, 3181–3189. [Google Scholar] [CrossRef]
- Okamoto, H. Ni-Zr (nickel-zirconium). J. Phase Equilibria Diffus. 2007, 28, 409–409. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I. Quantum Espresso: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395–502. [Google Scholar] [CrossRef] [PubMed]
- Garrity, K.F.; Bennett, J.W.; Rabe, K.M.; Vanderbilt, D. Pseudopotentials for high-throughput DFT calculations. Comput. Mater. Sci. 2014, 81, 446–452. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Methfessel, M.; Paxton, A.T. High-precision sampling for brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Takeshita, H.T.; Fujiwara, N.; Oishi, T.; Noréus, D.; Takeichi, N.; Kuriyama, N. Another unusual phenomenon for Zr7Ni10: Structural change in hydrogen solid solution and its conditions. J. Alloy. Compd. 2003, 360, 250–255. [Google Scholar] [CrossRef]
- Takeshita, H.; Kondo, S.; Miyamura, H.; Takeichi, N.; Kuriyama, N.; Oishi, T. Re-examination of Zr7Ni10 single-phase region. J. Alloy. Compd. 2004, 376, 268–274. [Google Scholar] [CrossRef]
- Kirkpatrick, M.; Smith, J.; Larsen, W. Structures of the intermediate phases Ni10Zr7 and Ni10Hf7. Acta Crystallogr. 1962, 15, 894–903. [Google Scholar] [CrossRef]
- Glimois, J.L.; Forey, P.; Feron, J.; Becle, C. Structural investigations of the pseudo-binary compounds Ni10−x CuxZr7. J. Less Common Met. 1981, 78, 45–50. [Google Scholar] [CrossRef]
- Joubert, J.M.; Cerný, R.; Yvon, K.; Latroche, M.; Percheron-Guégan, A. Zirconium–nickel, Zr7Ni10: Space group revision for the stoichiometric phase. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1997, 53, 1536–1538. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Eshelman, F.R.; Smith, J.F. The structure of Zr2Ni7. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1972, 28, 1594–1600. [Google Scholar] [CrossRef]
- Villars, P. Ni5Zr (ZrNi5) Crystal Structure. In Material Phases Data System (MPDS); Springer-Verlag GmbH: Heidelberg, Germany, 2014. [Google Scholar]
- Joubert, J.M.; Černý, R.; Yvon, K.; Latroche, M.; Percheron-Guégan, A. Refinement of the crystal structure of zirconium nickel, Zr8Ni21. Z. Kristallogr. New Cryst. Struct. 1998, 213, 227–228. [Google Scholar] [CrossRef]
- Villars, P. NiZr (ZrNi) Crystal Structure. In Material Phases Data System (MPDS); Springer-Verlag GmbH: Heidelberg, Germany, 2014. [Google Scholar]
- Nash, P.; Jayanth, C.S. The Ni-Zr (nickel-zirconium) system. Bull. Alloy Phase Diagr. 1984, 5, 144–148. [Google Scholar] [CrossRef]
- Young, K. Metal Hydrides. In Elsevier Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier B.V.: Waltham, MA, USA, 2013. [Google Scholar]
- Wong, D.F.; Young, K.; Ng, K.Y.S. First-principles study of structure, initial lattice expansion, and pressure-composition-temperature hysteresis for substituted LaNi5 and TiMn2 alloys. Model. Simul. Mater. Sci. Eng. 2016; submitted for publication. [Google Scholar]
- Schott, V.; Fähnle, M. Concentration of atomic defects in ordered compounds: Canonical and grandcanonical formalism. Phys. Status Solidi B 1997, 204, 617–624. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | This Work | References [28,29] | Reference [30] |
|---|---|---|---|
| Space group | Cmca | Aba2 | Cmca |
| a (Å) | 12.419 | 12.386 | 12.381 |
| b (Å) | 9.179 | 9.156 | 9.185 |
| c (Å) | 9.180 | 9.211 | 9.221 |
| Unit-cell volume (Å3) | 1046.5 | 1044.6 | 1048.6 |
| Defects | Ni-Rich | Stoichiometric | Zr-Rich |
|---|---|---|---|
| ΔHvZr (eV) | 2.12 | 2.22 | 2.33 |
| ΔHvNi (eV) | 1.10 | 1.03 | 0.95 |
| ΔHNiZr (eV) | 0.49 | 0.66 | 0.86 |
| ΔHZrNi (eV) | 0.80 | 0.63 | 0.44 |
| Parameters | Perfect Crystal | Zr Vacancy (4a) | Ni → Zr Anti-Site (8e) | Ni Vacancy (16g2) | Zr→Ni Anti-Site (8f2) |
|---|---|---|---|---|---|
| a (Å) | 12.418 | 12.334 | 12.365 | 12.379 | 12.437 |
| b (Å) | 9.179 | 9.152 | 9.135 | 9.172 | 9.224 |
| c (Å) | 9.180 | 9.139 | 9.152 | 9.162 | 9.230 |
| Unit-cell volume (Å3) | 1046.4 | 1031.6 | 1033.7 | 1040.3 | 1058.8 |
| Lattice Parameter | This Work | From Reference [32] |
|---|---|---|
| a (Å) | 4.677 | 4.698 |
| b (Å) | 8.239 | 8.235 |
| c (Å) | 12.176 | 12.193 |
| β (°) | 95.20 | 95.83 |
| Defects | Ni-Rich | Stoichiometric | Zr-Rich |
|---|---|---|---|
| ΔHvZr (eV) | 1.89 | 2.52 | 3.47 |
| ΔHvNi (eV) | 1.42 | 1.24 | 0.97 |
| ΔHNiZr (eV) | 0.48 | 1.29 | 2.52 |
| ΔHZrNi (eV) | 2.60 | 1.79 | 0.56 |
| Sample | Area | Zr | Ni | Al | Mn | B/A | Phase |
|---|---|---|---|---|---|---|---|
| Zr-rich Zr7Ni10 | 1 | 42.6 | 57.3 | 0.0 | 0.1 | 1.35 | Zr7Ni10 |
| 2 | 35.6 | 64.4 | 0.0 | 0.1 | 1.81 | Zr7Ni10 | |
| 3 | 38.4 | 59.4 | 0.0 | 2.2 | 1.60 | Zr7Ni10 | |
| 4 | 66.0 | 33.9 | 0.0 | 0.1 | 0.52 | Zr2Ni | |
| Ni-rich Zr7Ni10 | 1 | 40.4 | 59.6 | 0.0 | 0.0 | 1.48 | Zr7Ni10 |
| 2 | 39.9 | 60.1 | 0.0 | 0.0 | 1.51 | Zr7Ni10 | |
| 3 | 40.3 | 59.7 | 0.0 | 0.0 | 1.48 | Zr7Ni10 | |
| 4 | 40.2 | 59.8 | 0.0 | 0.0 | 1.49 | Zr7Ni10 | |
| 5 | 68.0 | 32.0 | 0.0 | 0.0 | 0.47 | Zr2Ni | |
| 6 | 59.9 | 40.1 | 0.0 | 0.0 | 0.67 | Zr2Ni |
| Parameter | Zr-Rich Zr7Ni10 | Ni-Rich Zr7Ni10 |
|---|---|---|
| a (Å) | 12.365 | 12.356 |
| b (Å) | 9.172 | 9.162 |
| c (Å) | 9.208 | 9.194 |
| Unit-cell volume (Å3) | 1044.2 | 1040.7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, D.F.; Young, K.-H.; Ouchi, T.; Ng, K.Y.S. First-Principles Point Defect Models for Zr7Ni10 and Zr2Ni7 Phases. Batteries 2016, 2, 23. https://doi.org/10.3390/batteries2030023
Wong DF, Young K-H, Ouchi T, Ng KYS. First-Principles Point Defect Models for Zr7Ni10 and Zr2Ni7 Phases. Batteries. 2016; 2(3):23. https://doi.org/10.3390/batteries2030023
Chicago/Turabian StyleWong, Diana F., Kwo-Hsiung Young, Taihei Ouchi, and K. Y. Simon Ng. 2016. "First-Principles Point Defect Models for Zr7Ni10 and Zr2Ni7 Phases" Batteries 2, no. 3: 23. https://doi.org/10.3390/batteries2030023
APA StyleWong, D. F., Young, K.-H., Ouchi, T., & Ng, K. Y. S. (2016). First-Principles Point Defect Models for Zr7Ni10 and Zr2Ni7 Phases. Batteries, 2(3), 23. https://doi.org/10.3390/batteries2030023