Electrochemical Performance and in Operando Charge Efficiency Measurements of Cu/Sn-Doped Nano Iron Electrodes

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Method
3. Results and Discussion
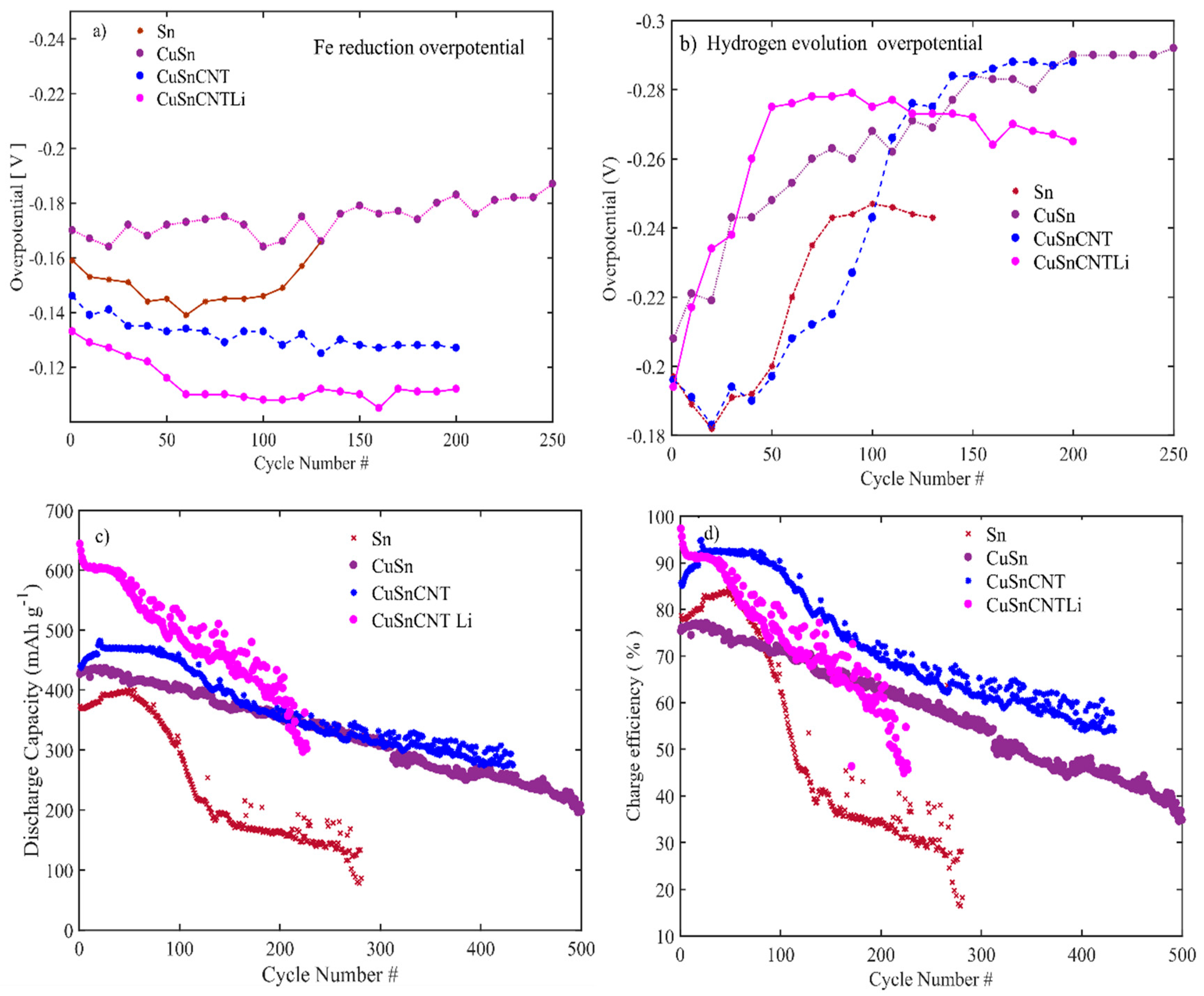
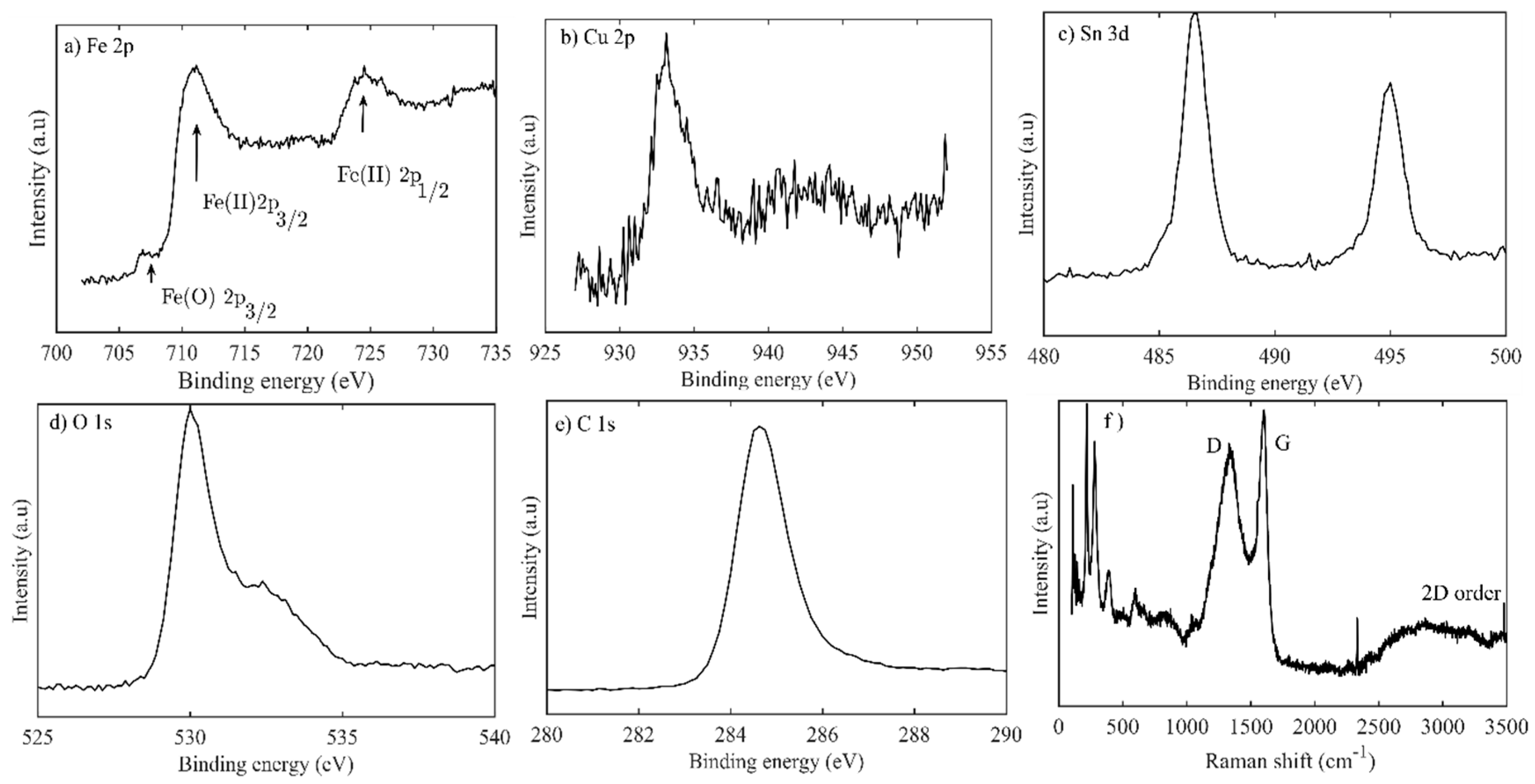
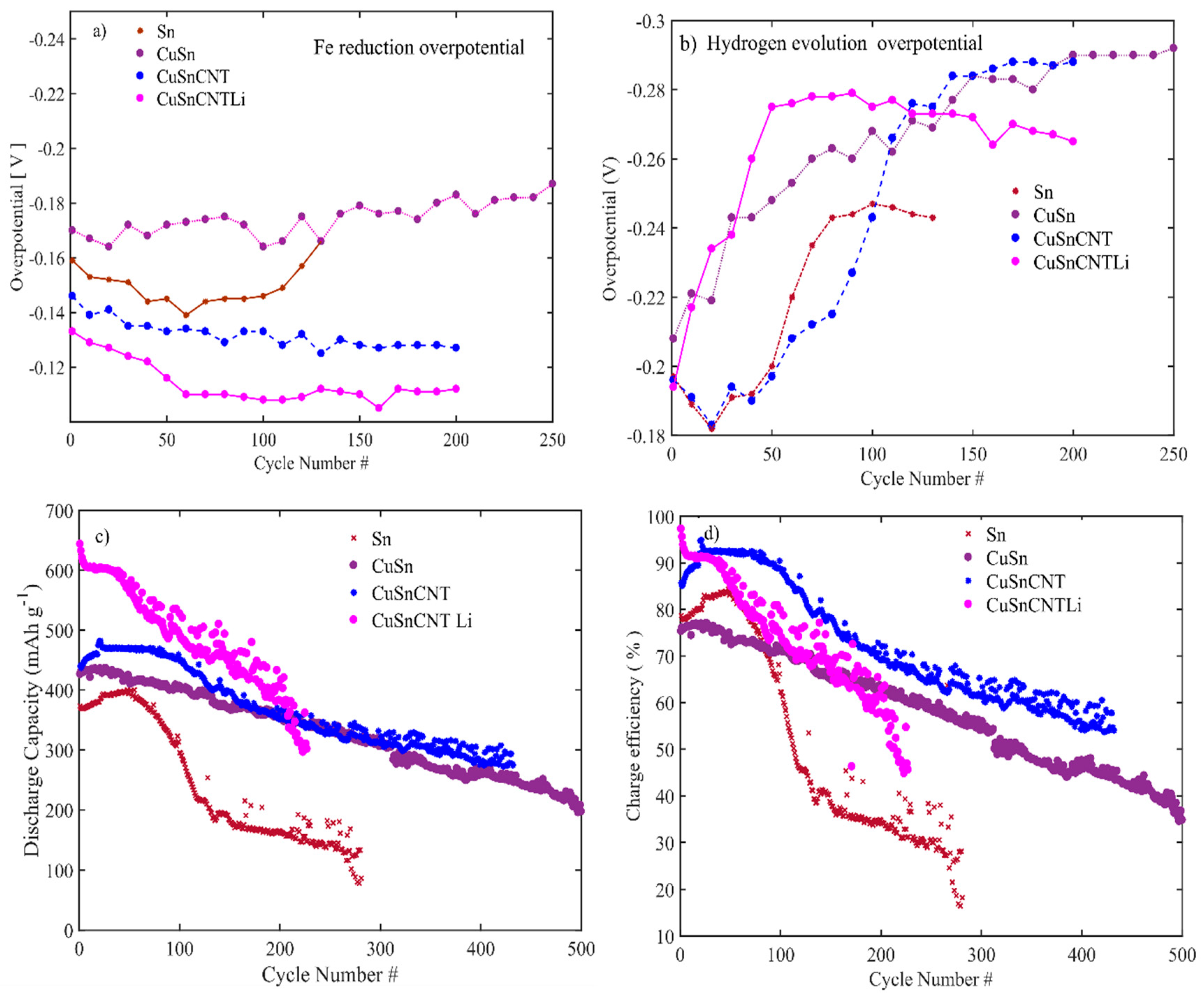
3.1. Material Characterizations
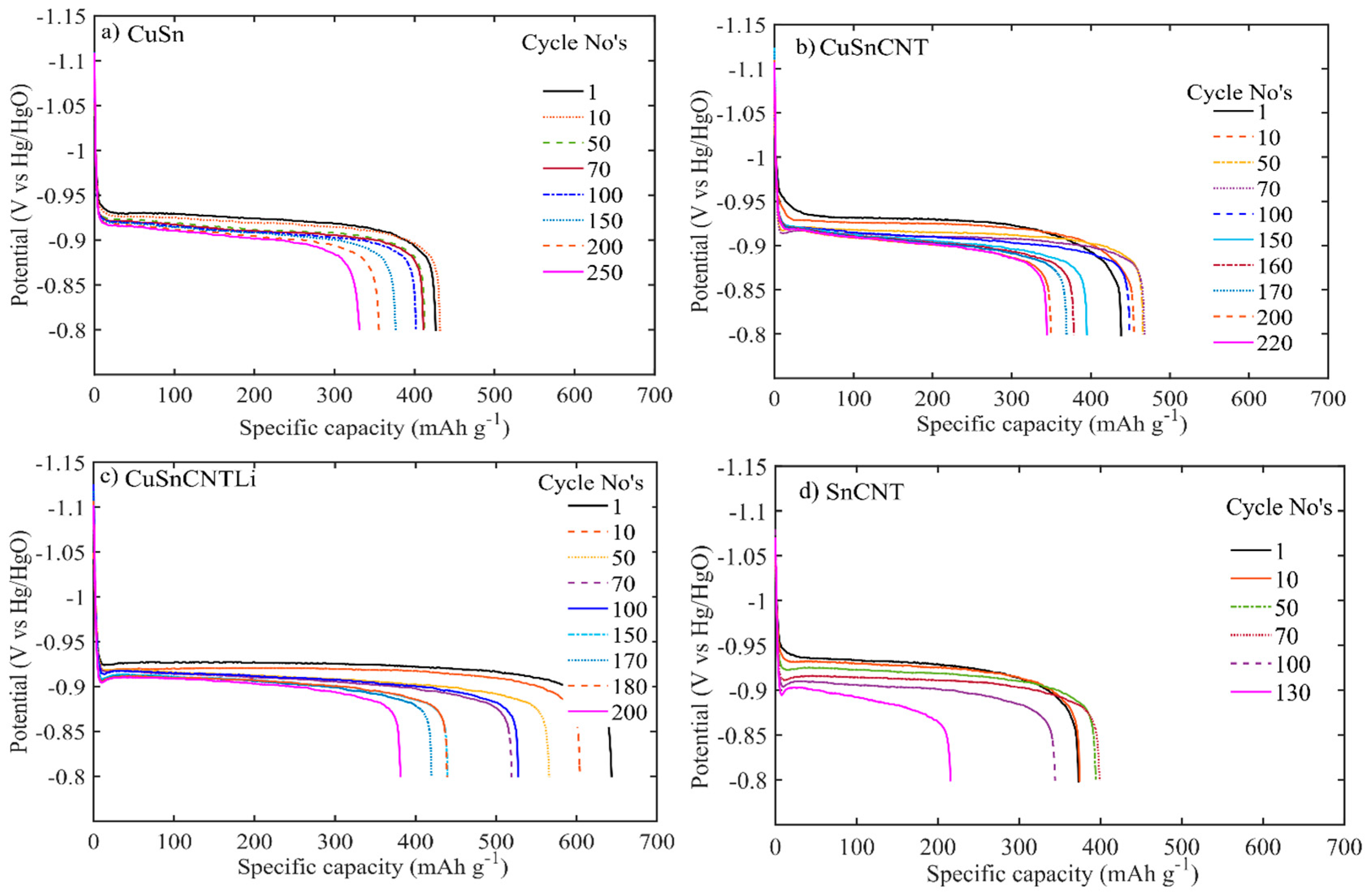
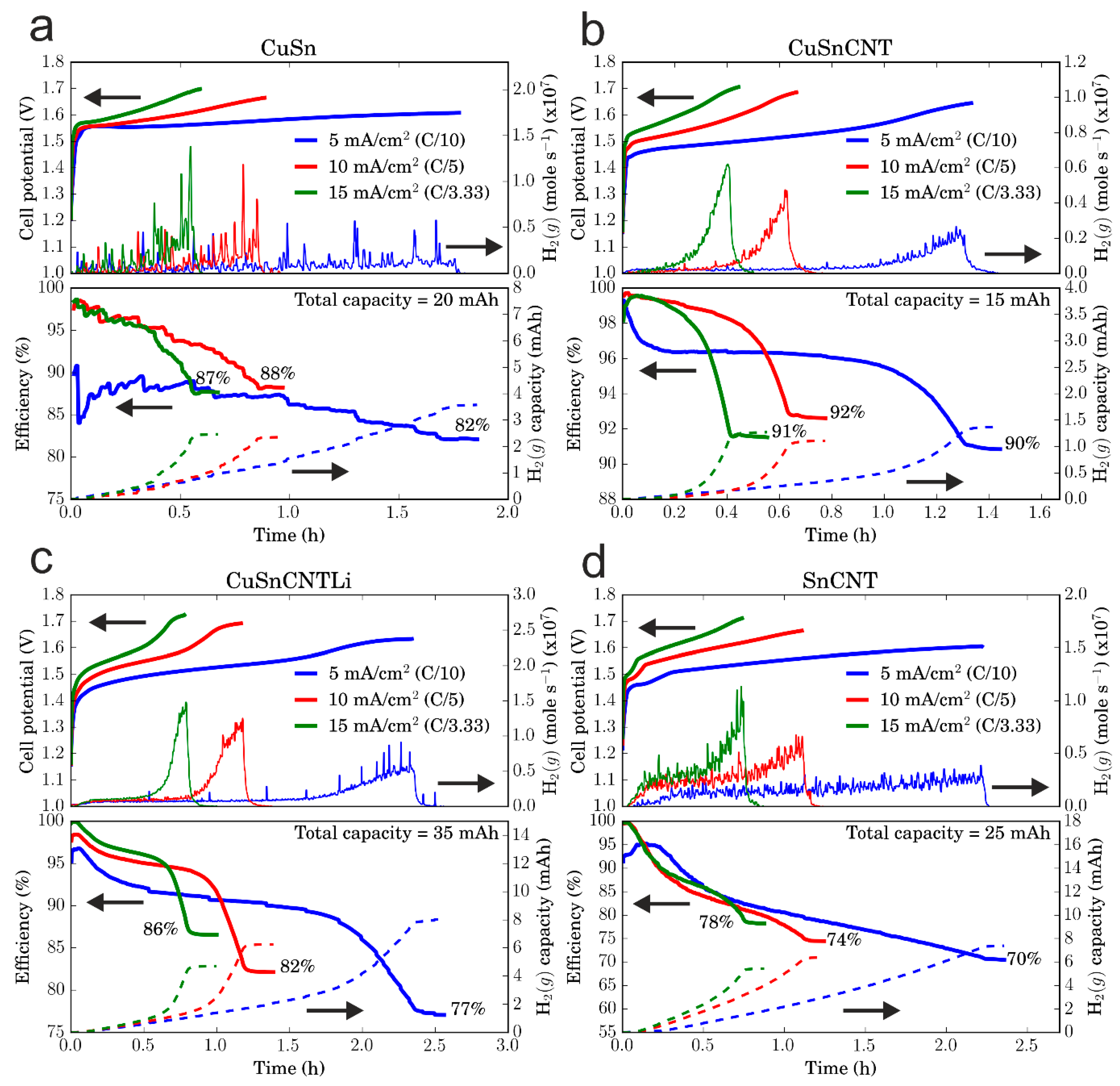
3.2. Electrochemical Performance of the Electrodes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matthews, H.D.; Kirsten, Z.; Reto, K.; Myles, R.A. Focus on cumulative emissions, global carbon budgets and the implications for climate mitigation targets. Environ. Res. Lett. 2018, 13, 010201. [Google Scholar] [CrossRef] [Green Version]
- Larcher, D.; Tarascon, J.M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, M.K.; Rathod, S.; Jaiswal, N.; Patil, S.; Shukla, A. The renaissance in redox flow batteries. J. Solid State Electrochem. 2016, 1–22. [Google Scholar] [CrossRef]
- Akinyele, D.O.; Rayudu, R.K. Review of energy storage technologies for sustainable power networks. Sustain. Energy Technol. Assess. 2014, 8, 74–91. [Google Scholar] [CrossRef]
- U.S. Department of Energy. Grid Energy Storage report. 2013. [Google Scholar]
- Öjefors, L.; Carlsson, L. An iron—Air vehicle battery. J. Power Sources 1978, 2, 287–296. [Google Scholar] [CrossRef]
- Narayanan, S.R.; Prakash, G.K.S.; Manohar, A.; Yang, B.; Malkhandi, S.; Kindler, A. Materials challenges and technical approaches for realizing inexpensive and robust iron–air batteries for large-scale energy storage. Solid State Ion. 2012, 216, 105–109. [Google Scholar] [CrossRef]
- Öjefors, L. Self-discharge of the alkaline iron electrode. Electrochim. Acta 1976, 21, 263–266. [Google Scholar] [CrossRef]
- Wang, H.; Liang, Y.; Gong, M.; Li, Y.; Chang, W.; Mefford, T.; Zhou, J.; Wang, J.; Regier, T.; Wei, F.; et al. An ultrafast nickel–iron battery from strongly coupled inorganic nanoparticle/nanocarbon hybrid materials. Nat. Commun. 2012, 3, 917. [Google Scholar] [CrossRef]
- Kao, C.-Y.; Tsai, Y.-R.; Chou, K.-S. Synthesis and characterization of the iron/copper composite as an electrode material for the rechargeable alkaline battery. J. Power Sources 2011, 196, 5746–5750. [Google Scholar] [CrossRef]
- Sundar Rajan, A.; Ravikumar, M.K.; Priolkar, K.R.; Sampath, S.; Shukla, A.K. Carbonyl-Iron Electrodes for Rechargeable-Iron Batteries. Electrochem. Energy Technol. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, M.K.; Rajan, A.S.; Sampath, S.; Priolkar, K.R.; Shukla, A.K. In Situ Crystallographic Probing on Ameliorating Effect of Sulfide Additives and Carbon Grafting in Iron Electrodes. J. Electrochem. Soc. 2015, 162, A2339–A2350. [Google Scholar] [CrossRef] [Green Version]
- Manohar, A.K.; Yang, C.; Malkhandi, S.; Prakash, G.K.S.; Narayanan, S.R. Enhancing the Performance of the Rechargeable Iron Electrode in Alkaline Batteries with Bismuth Oxide and Iron Sulfide Additives. J. Electrochem. Soc. 2013, 160, A2078–A2084. [Google Scholar] [CrossRef]
- Yang, C.; Manohar, A.K.; Narayanan, S.R. A High-Performance Sintered Iron Electrode for Rechargeable Alkaline Batteries to Enable Large-Scale Energy Storage. J. Electrochem. Soc. 2017, 164, A418–A429. [Google Scholar] [CrossRef] [Green Version]
- Manohar, A.K.; Yang, C.; Narayanan, S.R. The Role of Sulfide Additives in Achieving Long Cycle Life Rechargeable Iron Electrodes in Alkaline Batteries. J. Electrochem. Soc. 2015, 162, A1864–A1872. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Malkhandi, S.; Manohar, A.K.; Prakash, G.K.S.; Narayanan, S.R. Organo-sulfur molecules enable iron-based battery electrodes to meet the challenges of large-scale electrical energy storage. Energy Environ. Sci. 2014, 7, 2753–2763. [Google Scholar] [CrossRef]
- Hang, B.T.; Watanabe, T.; Egashira, M.; Watanabe, I.; Okada, S.; Yamaki, J.-I. The effect of additives on the electrochemical properties of Fe/C composite for Fe/air battery anode. J. Power Sources 2006, 155, 461–469. [Google Scholar] [CrossRef]
- Rajan, A.S.; Sampath, S.; Shukla, A.K. An in situ carbon-grafted alkaline iron electrode for iron-based accumulators. Energy Environ. Sci. 2014, 7, 1110–1116. [Google Scholar] [CrossRef]
- Balasubramanian, T.S.; Shukla, A.K. Effect of metal-sulfide additives on charge/discharge reactions of the alkaline iron electrode. J. Power Sources 1993, 41, 99–105. [Google Scholar] [CrossRef]
- Manohar, A.K.; Malkhandi, S.; Yang, B.; Yang, C.; Surya Prakash, G.K.; Narayanan, S.R. A High-Performance Rechargeable Iron Electrode for Large-Scale Battery-Based Energy Storage. J. Electrochem. Soc. 2012, 159, A1209–A1214. [Google Scholar] [CrossRef] [Green Version]
- Lei, D.; Lee, D.C.; Magasinski, A.; Zhao, E.; Steingart, D.; Yushin, G. Performance Enhancement and Side Reactions in Rechargeable Nickel-Iron Batteries with Nanostructured Electrodes. ACS Appl. Mater. Interfaces 2016, 8, 2088–2096. [Google Scholar] [CrossRef]
- Jiang, W.; Liang, F.; Wang, J.; Su, L.; Wu, Y.; Wang, L. Enhanced electrochemical performances of FeOx-graphene nanocomposites as anode materials for alkaline nickel-iron batteries. RSC Adv. 2014, 4, 15394–15399. [Google Scholar] [CrossRef]
- Ito, A.; Zhao, L.; Okada, S.; Yamaki, J.-I. Synthesis of nano-Fe3O4-loaded tubular carbon nanofibers and their application as negative electrodes for Fe/air batteries. J. Power Sources 2011, 196, 8154–8159. [Google Scholar] [CrossRef]
- Shangguan, E.; Guo, L.; Li, F.; Wang, Q.; Li, J.; Li, Q.; Chang, Z.; Yuan, X.-Z. FeS anchored reduced graphene oxide nanosheets as advanced anode material with superior high-rate performance for alkaline secondary batteries. J. Power Sources 2016, 327, 187–195. [Google Scholar] [CrossRef]
- Shangguan, E.; Li, F.; Li, J.; Chang, Z.; Li, Q.; Yuan, X.-Z.; Wang, H. FeS/C composite as high-performance anode material for alkaline nickel–iron rechargeable batteries. J. Power Sources 2015, 291, 29–39. [Google Scholar] [CrossRef]
- Hang, B.T.; Watanabe, T.; Eashira, M.; Watanabe, I.; Okada, S.; Yamaki, J.-I. Comparative Study of Fe2O3-Nanoloaded Carbon and Fe2O3-Nano/Carbon Mixed Composites for Iron-Air Battery Anodes. Electrochem. Solid-State Lett. 2005, 8, A476–A480. [Google Scholar] [CrossRef]
- Hang, B.T.; Watanabe, T.; Eashira, M.; Okada, S.; Yamaki, J.-I.; Hata, S.; Yoon, S.-H.; Mochida, I. The electrochemical properties of Fe2O3-loaded carbon electrodes for iron–air battery anodes. J. Power Sources 2005, 150, 261–271. [Google Scholar] [CrossRef]
- Hang, B.T.; Eashira, M.; Watanabe, I.; Okada, S.; Yamaki, J.-I.; Yoon, S.-H.; Mochida, I. The effect of carbon species on the properties of Fe/C composite for metal–air battery anode. J. Power Sources 2005, 143, 256–264. [Google Scholar] [CrossRef]
- Kao, C.Y.; Chou, K.S. Iron/carbon-black composite nanoparticles as an iron electrode material in a paste type rechargeable alkaline battery. J. Power Sources 2010, 195, 2399–2404. [Google Scholar] [CrossRef]
- Kim, T.; Ohata, Y.; Kim, J.; Rhee, C.K.; Miyawaki, J.; Yoon, S.-H. Fe nanoparticle entrained in tubular carbon nanofiber as an effective electrode material for metal–air batteries: A fundamental reason. Carbon 2014, 80, 698–707. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-C.; Chou, K.-S. Microstructure changes to iron nanoparticles during discharge/charge cycles. Electrochem. Commun. 2007, 9, 1907–1912. [Google Scholar] [CrossRef]
- Guo, C.X.; Li, C.M. Molecule-confined FeOx nanocrystals mounted on carbon as stable anode material for high energy density nickel-iron batteries. Nano Energy 2017, 42, 166–172. [Google Scholar] [CrossRef]
- Wu, X.; Wu, H.B.; Xiong, W.; Le, Z.; Sun, F.; Liu, F.; Chen, J.; Zhu, Z.; Lu, Y. Robust iron nanoparticles with graphitic shells for high-performance Ni-Fe battery. Nano Energy 2016, 30, 217–224. [Google Scholar] [CrossRef]
- Paulraj, A.R.; Kiros, Y.; Skårman, B.; Vidarsson, H. Core/Shell Structure Nano-Iron/Iron Carbide Electrodes for Rechargeable Alkaline Iron Batteries. J. Electrochem. Soc. 2017, 164, A1665–A1672. [Google Scholar] [CrossRef] [Green Version]
- Chamoun, M.; Skårman, B.; Vidarsson, H.; Smith, R.I.; Hull, S.; Lelis, M.; Milcius, D.; Noréus, D. Stannate Increases Hydrogen Evolution Overpotential on Rechargeable Alkaline Iron Electrodes. J. Electrochem. Soc. 2017, 164, A1251–A1257. [Google Scholar] [CrossRef] [Green Version]
- Kiros, Y.; Pirjamali, M.; Bursell, M. Oxygen reduction electrodes for electrolysis in chlor-alkali cells. Electrochim. Acta 2006, 51, 3346–3350. [Google Scholar] [CrossRef]
- Pirjamali, M.; Kiros, Y. Effects of carbon pretreatment for oxygen reduction in alkaline electrolyte. J. Power Sources 2002, 109, 446–451. [Google Scholar] [CrossRef]
- Svengren, H.; Chamoun, M.; Grins, J.; Johnsson, M. Water Splitting Catalysis Studied by using Real-Time Faradaic Efficiency Obtained through Coupled Electrolysis and Mass Spectrometry. ChemElectroChem 2018, 5, 44–50. [Google Scholar] [CrossRef]
- Li, F.; Pan, Y.; Wang, H.; Huang, X.; Zhang, Q.; Peng, Z.; Tang, Y. Core-Bishell Fe-Ni@Fe3O4@C Nanoparticles as an Advanced Anode for Rechargeable Nickel-Iron Battery. J. Electrochem. Soc. 2017, 164, A1333–A1338. [Google Scholar] [CrossRef]
- Wang, B.; Liu, T.; Liu, A.; Liu, G.; Wang, L.; Gao, T.; Wang, D.; Zhao, X.S. A Hierarchical Porous C@LiFePO4/Carbon Nanotubes Microsphere Composite for High-Rate Lithium-Ion Batteries: Combined Experimental and Theoretical Study. Adv. Energy Mater. 2016, 6, 1600426. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Zhou, C.; Wang, C.; Ba, X.; Li, Y.; Huang, X.; Liu, J. Carbon-Stabilized High-Capacity Ferroferric Oxide Nanorod Array for Flexible Solid-State Alkaline Battery–Supercapacitor Hybrid Device with High Environmental Suitability. Adv. Funct. Mater. 2015, 25, 5384–5394. [Google Scholar] [CrossRef]
- Li, F.; Shangguan, E.; Li, J.; Li, L.; Yang, J.; Chang, Z.; Li, Q.; Yuan, X.-Z.; Wang, H. Influence of annealing temperature on the structure and electrochemical performance of the Fe3O4 anode material for alkaline secondary batteries. Electrochim. Acta 2015, 178, 34–44. [Google Scholar] [CrossRef]
- Annamalai, A.; Shinde, P.S.; Jeon, T.H.; Lee, H.H.; Kim, H.G.; Choi, W.; Jang, J.S. Fabrication of superior α-Fe2O3 nanorod photoanodes through ex-situ Sn-doping for solar water splitting. Sol. Energy Mater. Sol. Cells 2016, 144, 247–255. [Google Scholar] [CrossRef]
- Xie, K.; Lu, Z.; Huang, H.; Lu, W.; Lai, Y.; Li, J.; Zhou, L.; Liu, Y. Iron supported C@Fe3O4 nanotube array: A new type of 3D anode with low-cost for high performance lithium-ion batteries. J. Mater. Chem. 2012, 22, 5560–5567. [Google Scholar] [CrossRef]
- Bokobza, L.; Bruneel, J.-L.; Couzi, M. Raman Spectra of Carbon-Based Materials (from Graphite to Carbon Black) and of Some Silicone Composites. C 2015, 1, 77–94. [Google Scholar] [CrossRef]
- Wu, K.-H.; Wang, D.-W.; Su, D.-S. An Extension to the Analytical Evaluation of the Oxygen Reduction Reaction Based On the Electrokinetics on a Rotating Ring–Disk Electrode. ChemElectroChem 2016. [Google Scholar] [CrossRef]
- Wang, B.; Xie, Y.; Liu, T.; Luo, H.; Wang, B.; Wang, C.; Wang, L.; Wang, D.; Dou, S.; Zhou, Y. LiFePO4 quantum-dots composite synthesized by a general microreactor strategy for ultra-high-rate lithium ion batteries. Nano Energy 2017, 42, 363–372. [Google Scholar] [CrossRef]
- Wang, B.; Al Abdulla, W.; Wang, D.; Zhao, X.S. A three-dimensional porous LiFePO4 cathode material modified with a nitrogen-doped graphene aerogel for high-power lithium ion batteries. Energy Environ. Sci. 2015, 8, 869–875. [Google Scholar] [CrossRef]
- Hansal, W.E.G.; Halmdienst, M.; Hansal, S.; Boussaboua, I.; Darchen, A. Influence of pulse plating parameters on morphology and hardness of pure tin deposit. Trans. IMF 2008, 86, 115–121. [Google Scholar] [CrossRef]
- Bani Hashemi, A.; La Mantia, F. Cell Design for Electrochemical Characterizations of Metal-Ion Batteries in Organic and Aqueous Electrolyte. Anal. Chem. 2016, 88, 7916–7920. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Sample ID | Fe loading (mg cm−2) |
|---|---|---|
| 2 wt. % Cu + 2 wt. % Sn | CuSn | 29 |
| 2 wt. % Cu + 2 wt. % Sn + 0.1 wt. % CNT | CuSnCNT | 27 |
| 2 wt. % Cu + 2 wt. % Sn + 0.1 wt. % CNT + 0.65 M of LiOH | CuSnCNTLi | 27 |
| 4 wt. % Sn + 0.1 wt. % CNT | SnCNT | 22 |
| Element | ASF Corrected XPS Peak Intensity |
|---|---|
| Fe | 40 |
| Cu | 1 |
| O | 95 |
| Sn | 20 |
| Electrode | Initial Discharge Capacity (mAh g−1) | Initial Charge Efficiency (%) |
|---|---|---|
| CuSn | 434 | 82.6 |
| CuSnCNT | 470 | 93.0 |
| CuSnCNTLi | 603 | 91.3 |
| SnCNT | 378 | 76.7 |
| Sample | ||
|---|---|---|
| CuSn | 82/88/87% | 81/88/88% |
| CuSnCNT | 90/92/91% | 93/92/90% |
| CuSnCNTLi | 77/82/86% | 77/83/88% |
| SnCNT | 70/74/78% | 70/76/80% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulraj, A.R.; Kiros, Y.; Chamoun, M.; Svengren, H.; Noréus, D.; Göthelid, M.; Skårman, B.; Vidarsson, H.; Johansson, M.B. Electrochemical Performance and in Operando Charge Efficiency Measurements of Cu/Sn-Doped Nano Iron Electrodes. Batteries 2019, 5, 1. https://doi.org/10.3390/batteries5010001
Paulraj AR, Kiros Y, Chamoun M, Svengren H, Noréus D, Göthelid M, Skårman B, Vidarsson H, Johansson MB. Electrochemical Performance and in Operando Charge Efficiency Measurements of Cu/Sn-Doped Nano Iron Electrodes. Batteries. 2019; 5(1):1. https://doi.org/10.3390/batteries5010001
Chicago/Turabian StylePaulraj, Alagar Raj, Yohannes Kiros, Mylad Chamoun, Henrik Svengren, Dag Noréus, Mats Göthelid, Björn Skårman, Hilmar Vidarsson, and Malin B. Johansson. 2019. "Electrochemical Performance and in Operando Charge Efficiency Measurements of Cu/Sn-Doped Nano Iron Electrodes" Batteries 5, no. 1: 1. https://doi.org/10.3390/batteries5010001