
Optimized Lambda Exonuclease Digestion or Purification Using Streptavidin-Coated Beads: Which One Is Best for Successful DNA Aptamer Selection?
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. ssDNA Library and Primers
2.2. Amplification of ssDNA Library
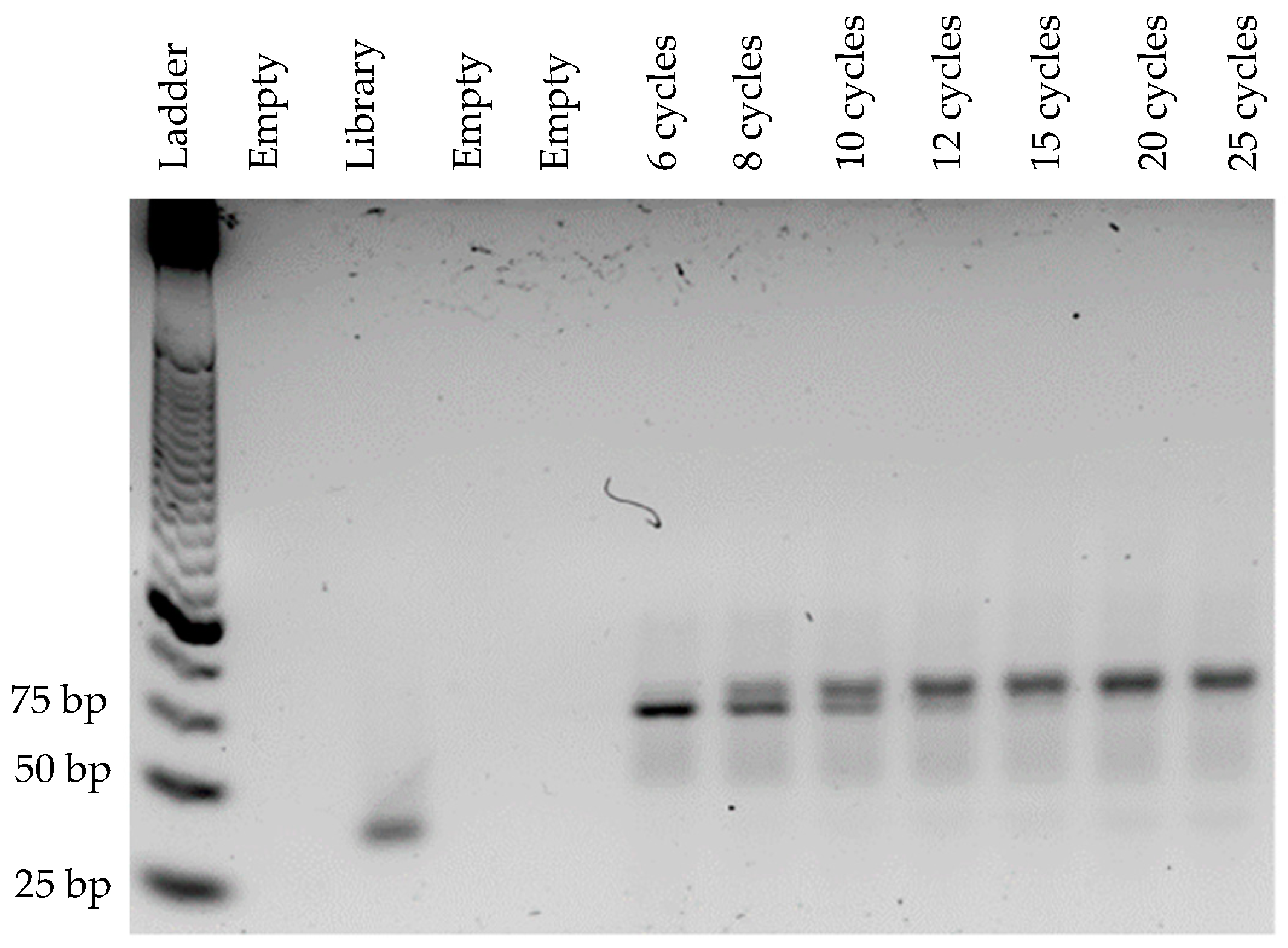
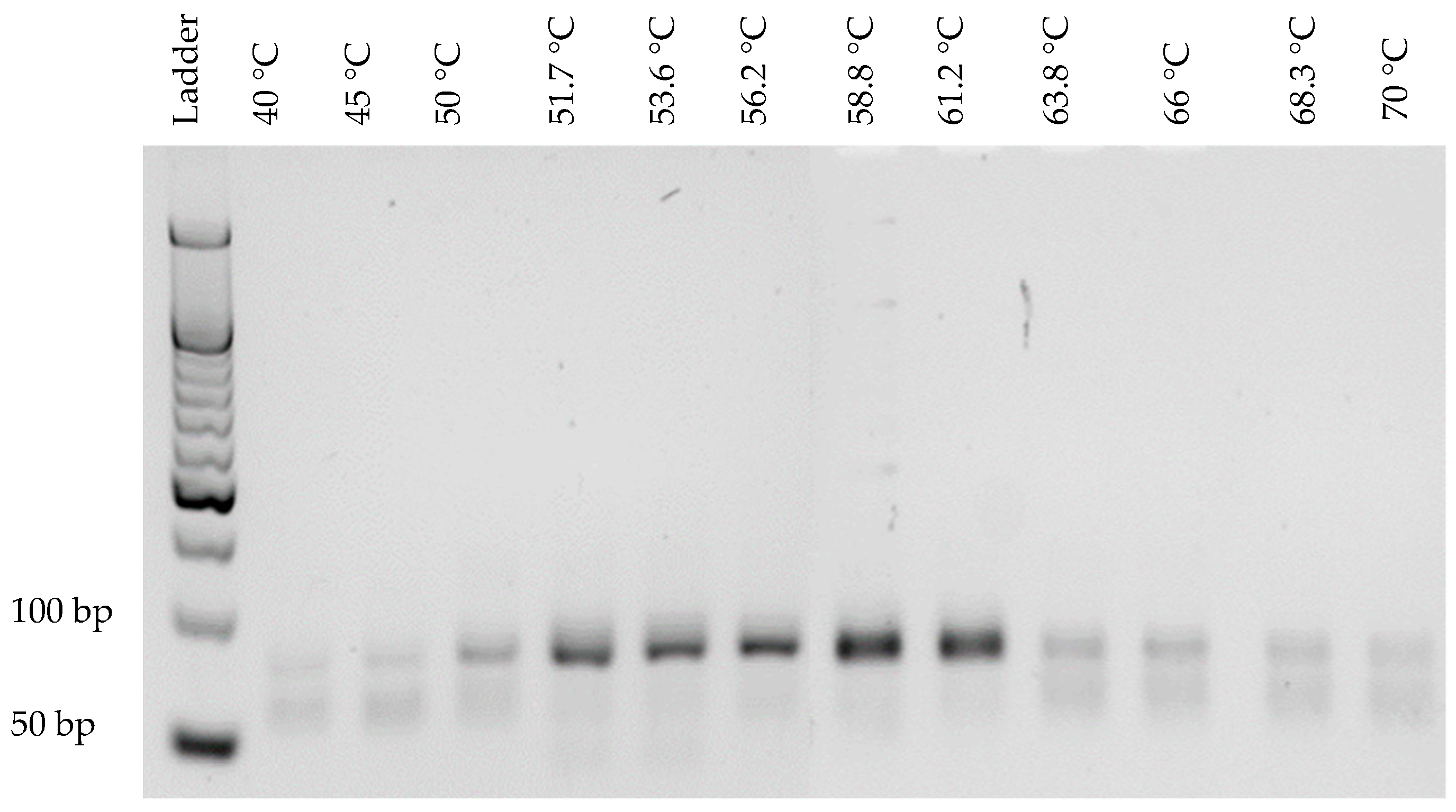
2.2.1. Optimization of PCR Conditions
2.2.2. Large-Scale PCR Amplification
2.3. Optimization of Capture on Streptavidin-Coated Magnetic Beads
2.4. Optimization of Lambda Exonuclease Digestion
2.5. Comparison of ssDNA Generation Methods
2.5.1. Purification of ssDNA
2.5.2. ssDNA Quantification Techniques
2.5.3. ssDNA Recovery
2.5.4. Statistical Analysis
3. Results
3.1. Optimization of PCR Conditions
3.2. Optimization of Capture on Streptavidin-Coated Magnetic Beads
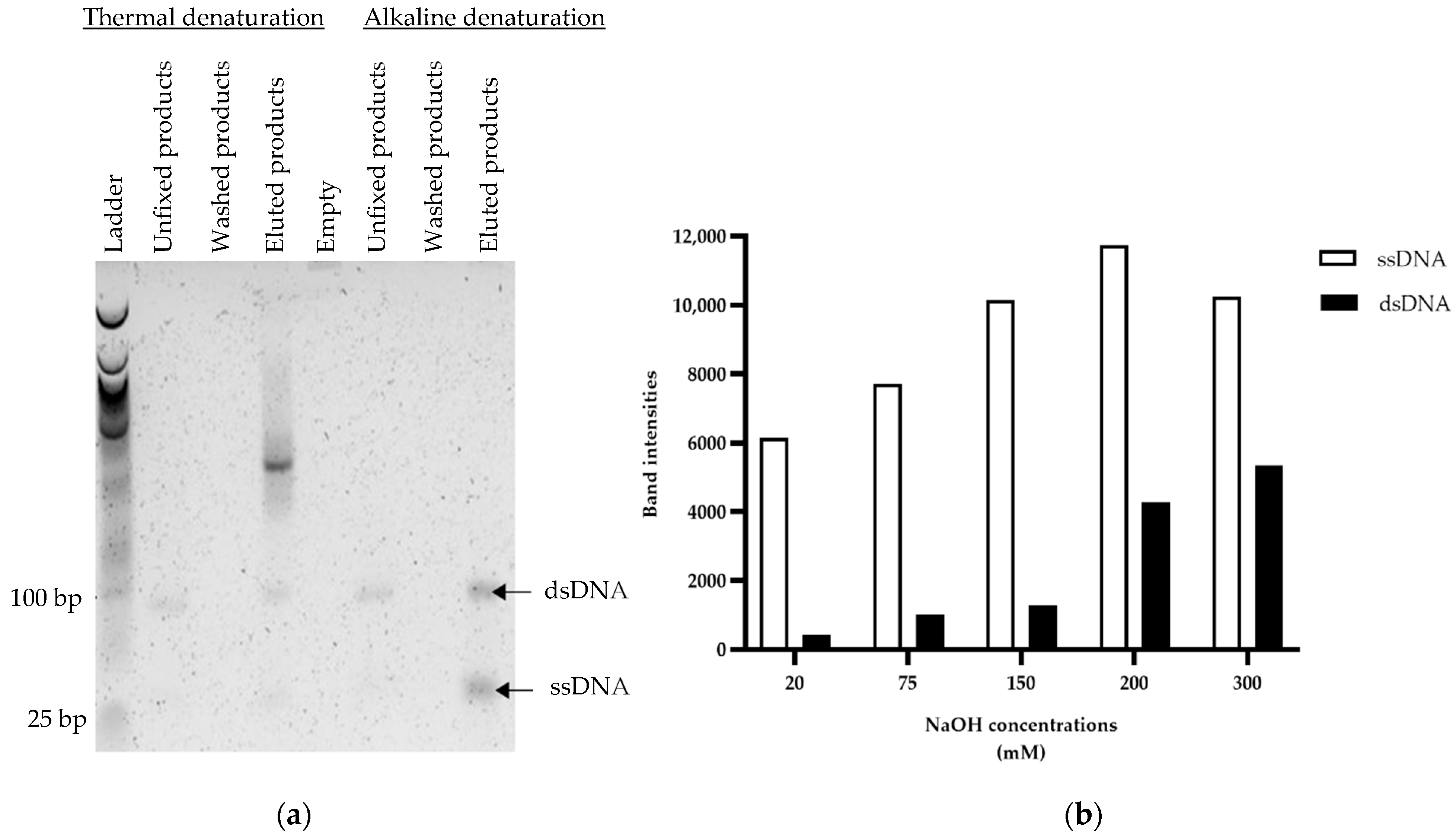
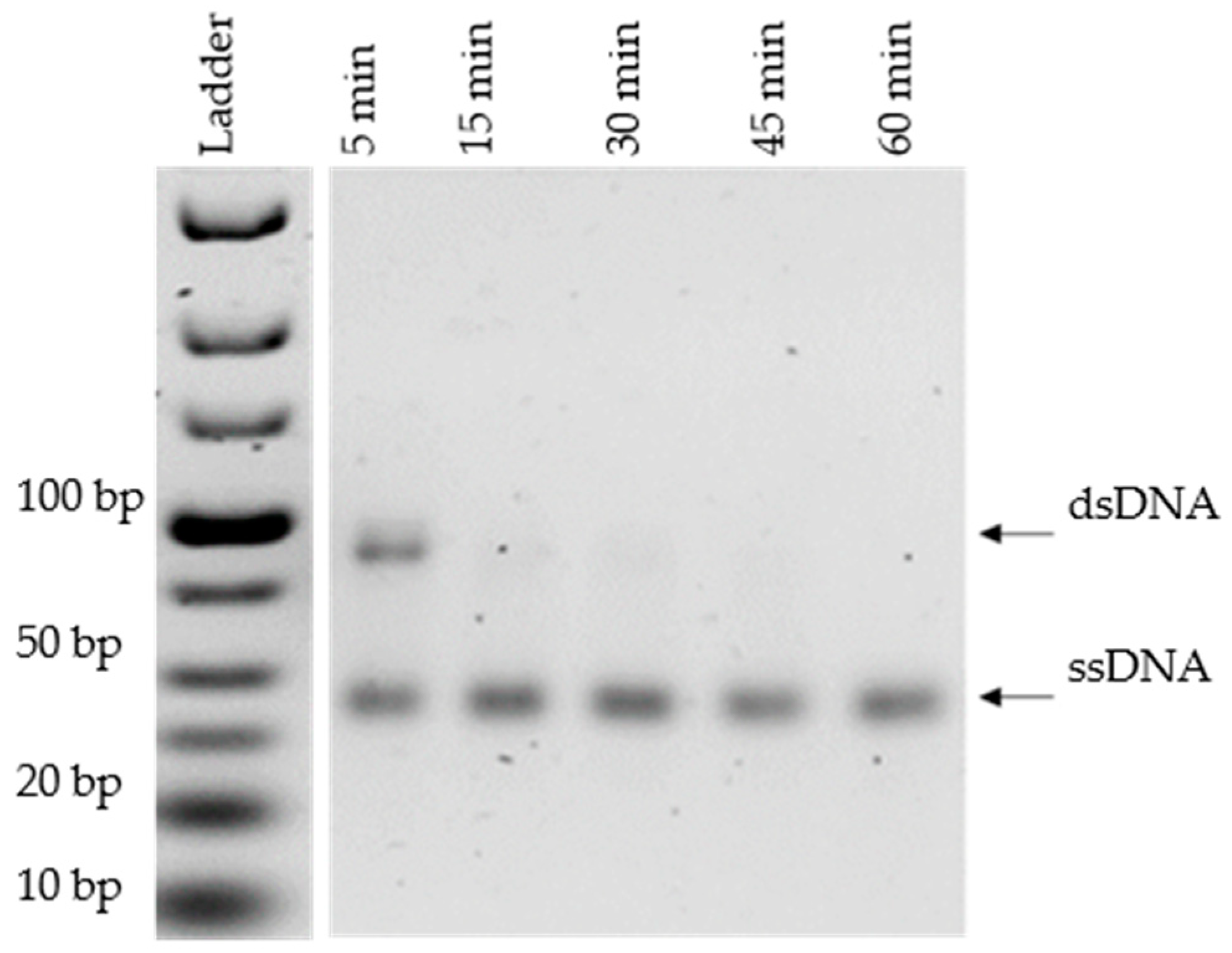
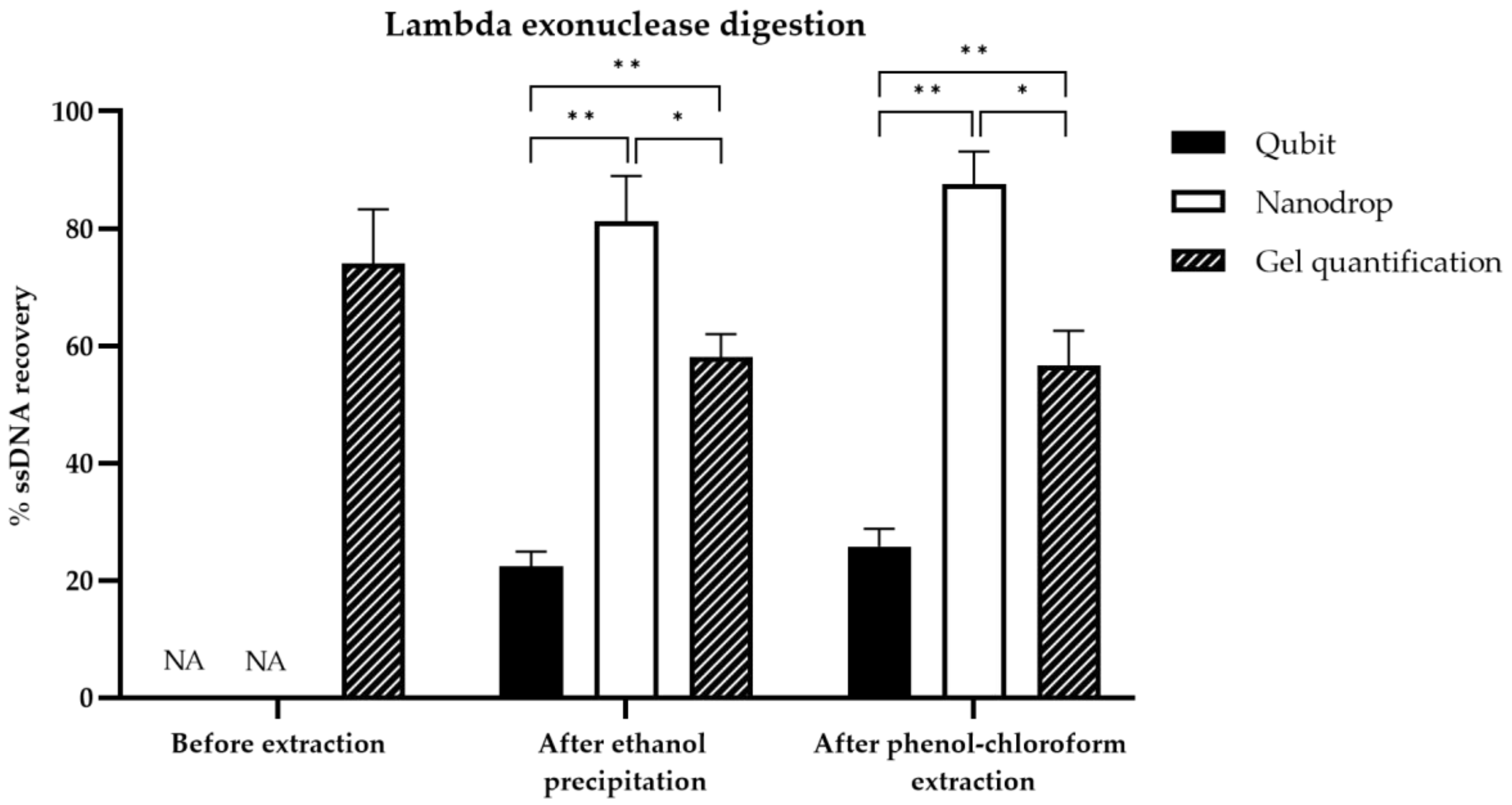
3.3. Optimization of Lambda Exonuclease Digestion
3.4. Comparison of ssDNA Quantification Techniques
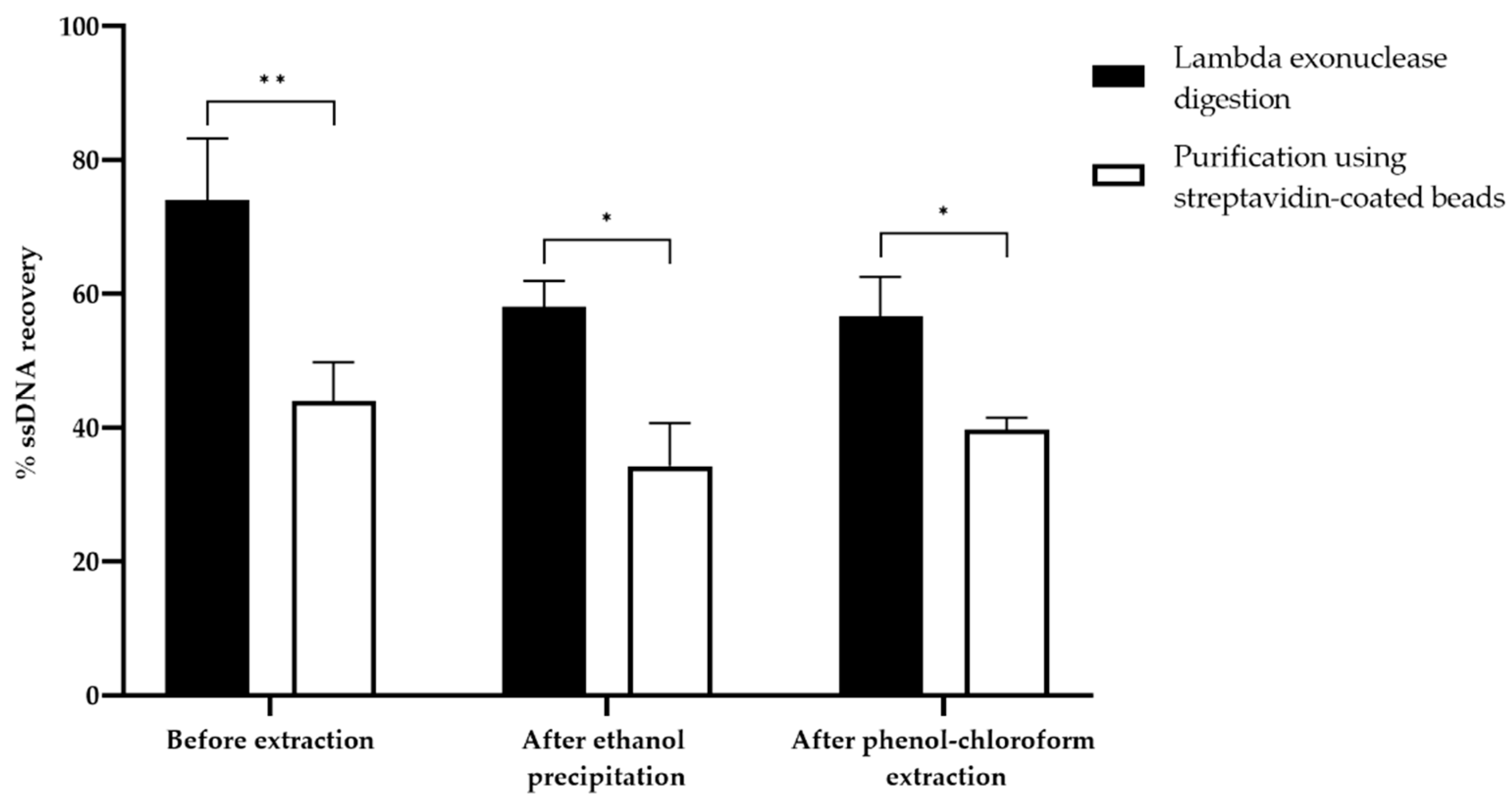
3.5. Comparison of ssDNA Generation Methods
3.5.1. Quantitative Evaluation
3.5.2. Qualitative Evaluation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yin, W.; AlShamaileh, H.; Zhang, Y.; Tran, P.H.-L.; Nguyen, T.N.-G.; Li, Y.; Chen, K.; Sun, M.; Hou, Y.; et al. A Detailed Protein-SELEX Protocol Allowing Visual Assessments of Individual Steps for a High Success Rate. Hum. Gene Ther. Methods 2019, 30, 1–16. [Google Scholar] [CrossRef]
- Ohuchi, S. Cell-SELEX Technology. BioResearch Open Access 2012, 1, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Marton, S.; Cleto, F.; Krieger, M.A.; Cardoso, J. Isolation of an Aptamer that Binds Specifically to E. coli. PLoS ONE 2016, 11, e0153637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bala, J.; Chinnapaiyan, S.; Dutta, R.K.; Unwalla, H. Aptamers in HIV research diagnosis and therapy. RNA Biol. 2018, 15, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Röthlisberger, P.; Hollenstein, M. Aptamer chemistry. Adv. Drug Deliv. Rev. 2018, 134, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lai, B.S.; Juhas, M. Recent Advances in Aptamer Discovery and Applications. Molecules 2019, 24, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordasht, H.K.; Hasanzadeh, M. Aptamer based recognition of cancer cells: Recent progress and challenges in bioanalysis. Talanta 2020, 220, 121436. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef]
- Park, K.S. Nucleic acid aptamer-based methods for diagnosis of infections. Biosens. Bioelectron. 2018, 102, 179–188. [Google Scholar] [CrossRef]
- Dougherty, C.A.; Cai, W.; Hong, H. Applications of Aptamers in Targeted Imaging: State of the Art. Curr. Top. Med. Chem. 2015, 15, 1138–1152. [Google Scholar] [CrossRef]
- Famulok, M.; Mayer, G. Aptamers and SELEX in Chemistry & Biology. Chem. Biol. 2014, 21, 1055–1058. [Google Scholar]
- Oteng, E.K.; Gu, W.; McKeague, M. High-efficiency enrichment enables identification of aptamers to circulating Plasmodium falciparum-infected erythrocytes. Sci. Rep. 2020, 10, 9706. [Google Scholar] [CrossRef]
- Citartan, M.; Tang, T.-H.; Tominaga, J.; Tan, S.-C.; Gopinath, S.C.B. Single-stranded DNA (ssDNA) production in DNA aptamer generation. Analyst 2012, 137, 1307–1315. [Google Scholar]
- Komarova, N.; Kuznetsov, A. Inside the Black Box: What Makes SELEX Better? Molecules 2019, 24, 3598. [Google Scholar] [CrossRef] [Green Version]
- Yeoh, T.S.; Anna, A.; Tang, T.-H.; Citartan, M. Development of an optimization pipeline of asymmetric PCR towards the generation of DNA aptamers: A guide for beginners. World J. Microbiol. Biotechnol. 2022, 38, 31. [Google Scholar] [CrossRef]
- Tabarzad, M.; Kazemi, B.; Vahidi, H.; Aboofazeli, R.; Shahhosseini, S.; Nafissi-Varcheh, N. Challenges to Design and Develop of DNA Aptamers for Protein Targets. I. Optimization of Asymmetric PCR for Generation of a Single Stranded DNA Library. Iran. J. Pharm. Res. IJPR 2014, 13, 133–141. [Google Scholar]
- Heiat, M.; Ranjbar, R.; Latifi, A.M.; Rasaee, M.J.; Farnoosh, G. Essential strategies to optimize asymmetric PCR conditions as a reliable method to generate large amount of ssDNA aptamers. Biotechnol. Appl. Biochem. 2017, 64, 541–548. [Google Scholar] [CrossRef]
- Avci-Adali, M.; Paul, A.; Wilhelm, N.; Ziemer, G.; Wendel, H.P. Upgrading SELEX technology by using lambda exonuclease digestion for single-stranded DNA generation. Molecules 2009, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, H.; Zhou, H.; Wu, F.; Su, Y.; Liang, Y.; Zhou, D. Indirect purification method provides high yield and quality ssDNA sublibrary for potential aptamer selection. Anal. Biochem. 2015, 476, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Avci-Adali, M.; Ziemer, G.; Wendel, H.P. Streptavidin-coated magnetic beads for DNA strand separation implicate a multitude of problems during cell-SELEX. Oligonucleotides 2019, 19, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Citartan, M.; Tang, T.-H.; Tan, S.C.; Gopinath, S.C.B. Conditions optimized for the preparation of single-stranded DNA (ssDNA) employing lambda exonuclease digestion in generating DNA aptamer. World J. Microbiol. Biotechnol. 2011, 27, 1167–1173. [Google Scholar] [CrossRef]
- Svobodová, M.; Pinto, A.; Nadal, P.; O’ Sullivan, C.K. Comparison of different methods for generation of single-stranded DNA for SELEX processes. Anal. Bioanal. Chem. 2012, 404, 835–842. [Google Scholar] [CrossRef] [Green Version]
- Civit, L.; Fragoso, A.; O’Sullivan, C.K. Evaluation of techniques for generation of single-stranded DNA for quantitative detection. Anal. Biochem. 2012, 431, 132–138. [Google Scholar] [CrossRef]
- Kilili, G.K.; Tilton, L.; Karbiwnyk, C.M. NaOH concentration and streptavidin bead type are key factors for optimal DNA aptamer strand separation and isolation. BioTechniques 2016, 61, 114–116. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, A.; Liu, J.; Dixon, B.; Zargar, B.; Sattar, S.A. Development and application of DNA-aptamer-coupled magnetic beads and aptasensors for the detection of Cryptosporidium parvum oocysts in drinking and recreational water resources. Can. J. Microbiol. 2019, 65, 851–857. [Google Scholar] [CrossRef]
- Dynabeads Streptavidin FAQ—FR. Available online: https://www.thermofisher.com/us/en/home/brands/product-brand/dynal/streptavidin-coupled-dynabeads/dynabeads-streptavidin.html (accessed on 23 October 2022).
- Moore, D.; Dowhan, D. Purification and concentration of DNA from aqueous solutions. Curr. Protoc. Mol. Biol. 2002, 59, 1–2. [Google Scholar] [CrossRef]
- Tolle, F.; Wilke, J.; Wengel, J.; Mayer, G. By-Product Formation in Repetitive PCR Amplification of DNA Libraries during SELEX. PLoS ONE 2014, 9, e114693. [Google Scholar] [CrossRef] [Green Version]
- Tominaga, J.; Saini, R.; Hoe, C.-H.; Tang, T.-H.; Tan, S.-C.; Citartan, M.; Gopinath, S.C.B. Asymmetric PCR for good quality ssDNA generation towards DNA aptamer production. Songklanakarin J. Sci. Technol. SJST 2012, 34, 125–131. [Google Scholar]
- Wilson, R. Preparation of single-stranded DNA from PCR products with streptavidin magnetic beads. Nucleic Acid Ther. 2011, 21, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Lambda Exonuclease NEB. Available online: https://www.neb.com/products/m0262-lambda-exonuclease#Product%20Information_Product%20Notes (accessed on 23 October 2022).
- Sapia, R.J.; Campbell, C.; Reed, A.J.; Tsvetkov, V.B.; Gerasimova, Y.V. Interaction of GelRedTM with single-stranded DNA oligonucleotides: Preferential binding to thymine-rich sequences. Dye. Pigment. 2021, 188, 109209. [Google Scholar] [CrossRef]
- Nakayama, Y.; Yamaguchi, H.; Einaga, N.; Esumi, M. Pitfalls of DNA Quantification Using DNA-Binding Fluorescent Dyes and Suggested Solutions. PLoS ONE 2016, 11, e0150528. [Google Scholar] [CrossRef] [PubMed]
- Koetsier, G.; Cantor, E. A Practical Guide to Analyzing Nucleic Acid Concentration and Purity with Microvolume Spectrophotometers NEB 2019. Available online: https://www.science.smith.edu/cmbs/wp-content/uploads/sites/36/2020/01/A-Practical-Guide-to-Analyzing-Nucleic-Acid-Concentration-and-Purity-with-Microvolume-Spectrophotometers.pdf (accessed on 7 July 2022).
- Liang, C.; Li, D.; Zhang, G.; Li, H.; Shao, N.; Liang, Z.; Zhang, L.; Lu, A.; Zhang, G. Comparison of the methods for generating single-stranded DNA in SELEX. Analyst 2015, 140, 3439–3444. [Google Scholar] [CrossRef]
- Lambda Exonuclease (10 U/µL). Available online: https://www.thermofisher.com/order/catalog/product/EN0561 (accessed on 23 October 2022).
- Nosaz, Z.; Rasoulinejad, S.; Mousavi Gargari, S.L. Development of a DNA aptamer to detect Brucella abortus and Brucella melitensis through cell SELEX. Iran. J. Vet. Res. 2020, 21, 294–300. [Google Scholar]
- Bitaraf, F.S.; Rasooli, I.; Mousavi Gargari, S.L. DNA aptamers for the detection of Haemophilus influenzae type b by cell SELEX. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 503–510. [Google Scholar] [CrossRef]
- Bibby, D.F.; Gill, A.C.; Kirby, L.; Farquhar, C.F.; Bruce, M.E.; Garson, J.A. Application of a novel in vitro selection technique to isolate and characterise high affinity DNA aptamers binding mammalian prion proteins. J. Virol. Methods 2018, 151, 107–115. [Google Scholar] [CrossRef]
- Veneziano, R.; Shepherd, T.R.; Ratanalert, S.; Bellou, L.; Tao, C.; Bathe, M. In vitro synthesis of gene-length single-stranded DNA. Sci. Rep. 2018, 8, 6548. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Dortz, L.L.; Rouxel, C.; Leroy, Q.; Brosseau, N.; Boulouis, H.-J.; Haddad, N.; Lagrée, A.-C.; Deshuillers, P.L. Optimized Lambda Exonuclease Digestion or Purification Using Streptavidin-Coated Beads: Which One Is Best for Successful DNA Aptamer Selection? Methods Protoc. 2022, 5, 89. https://doi.org/10.3390/mps5060089
Le Dortz LL, Rouxel C, Leroy Q, Brosseau N, Boulouis H-J, Haddad N, Lagrée A-C, Deshuillers PL. Optimized Lambda Exonuclease Digestion or Purification Using Streptavidin-Coated Beads: Which One Is Best for Successful DNA Aptamer Selection? Methods and Protocols. 2022; 5(6):89. https://doi.org/10.3390/mps5060089
Chicago/Turabian StyleLe Dortz, Lisa Lucie, Clotilde Rouxel, Quentin Leroy, Noah Brosseau, Henri-Jean Boulouis, Nadia Haddad, Anne-Claire Lagrée, and Pierre Lucien Deshuillers. 2022. "Optimized Lambda Exonuclease Digestion or Purification Using Streptavidin-Coated Beads: Which One Is Best for Successful DNA Aptamer Selection?" Methods and Protocols 5, no. 6: 89. https://doi.org/10.3390/mps5060089