
Obtaining Boron Carbide and Nitride Matrix Nanocomposites for Neutron-Shielding and Therapy Applications
 , , ,
, , ,
Abstract
:1. Introduction
2. Experimental—Materials and Equipment
2.1. Materials
2.2. Technological Facilities
2.3. Measuring Apparatus
3. Boron Carbide–Tungsten Layered Composites
3.1. Available Methods of Obtaining Tungsten Layers
3.2. Developed Methods of Obtaining Boron Carbide–Tungsten Sandwich Structures
3.2.1. Synthesis of Peroxotungstic Acid
3.2.2. Tungsten Deposition on Boron Carbide Surface
3.2.3. Compaction of Boron Carbide and Tungsten Powders
3.2.4. Compaction of Boron Carbide Powder with Tungsten Foil
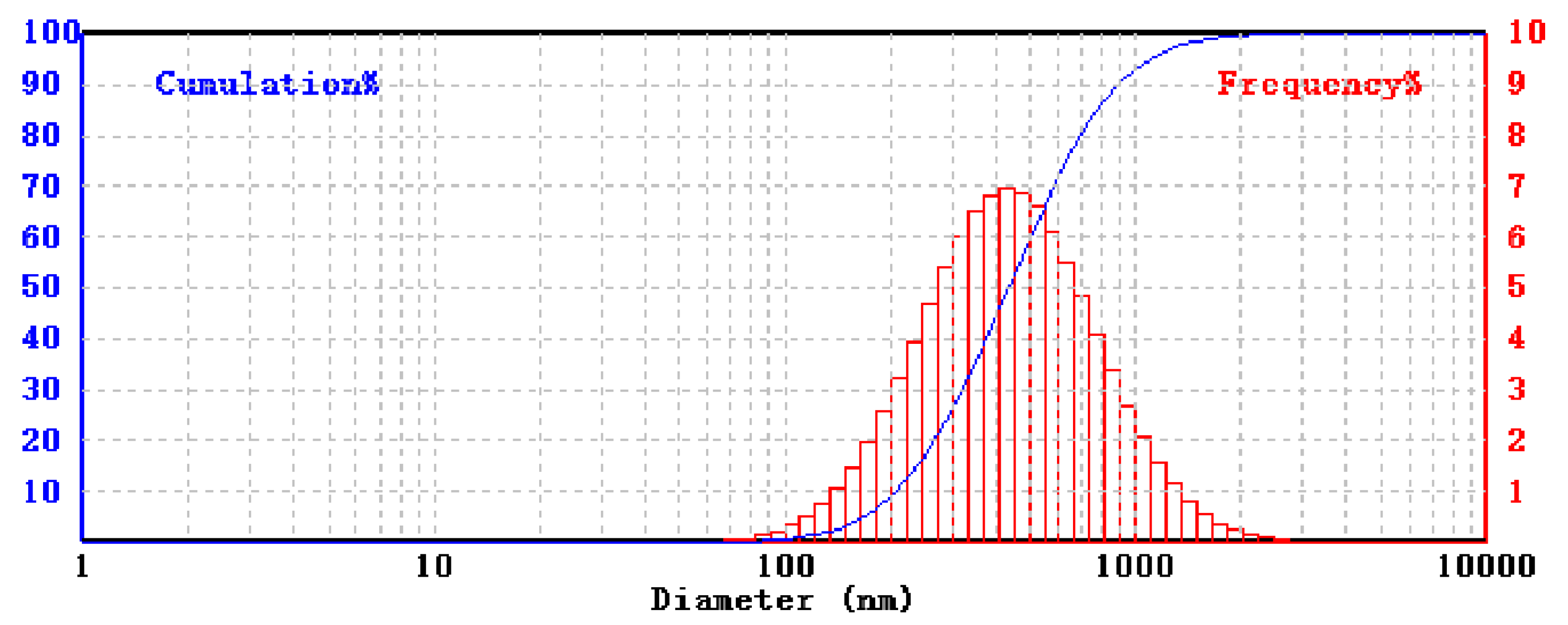
3.3. Characterization of Boron Carbide–Tungsten Sandwich Structures
3.4. Further Analysis of Boron Carbide–Tungsten Composites
3.4.1. B–W Systems
3.4.2. C–W Systems
3.4.3. B–C–W Systems
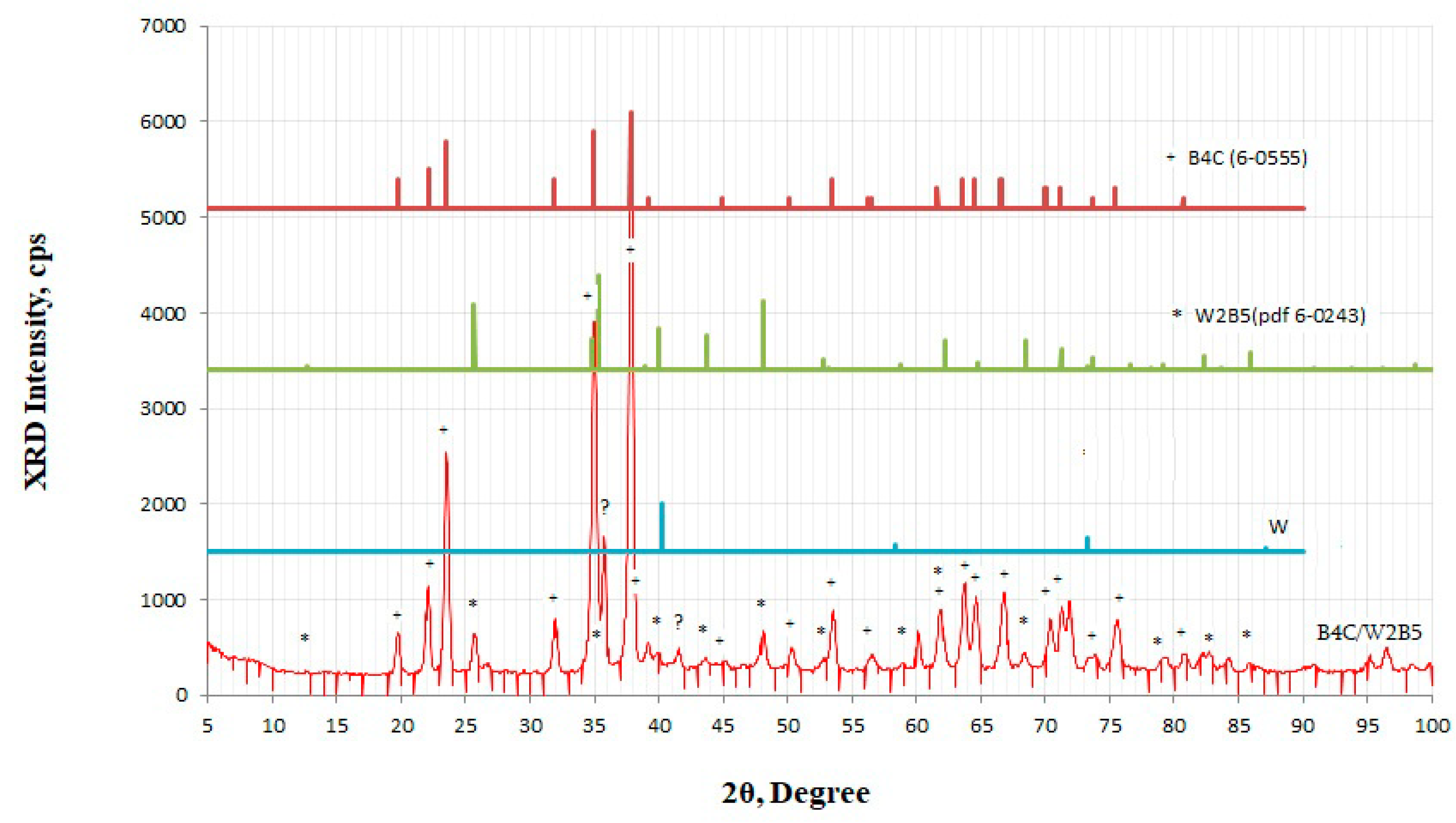
3.4.4. B4C–W Systems
3.4.5. B4C–WB2 Systems
3.4.6. B4C–W2B5 Systems
4. Hexagonal Boron Nitride–Iron and Magnetite Composites
4.1. Test Samples Preparation
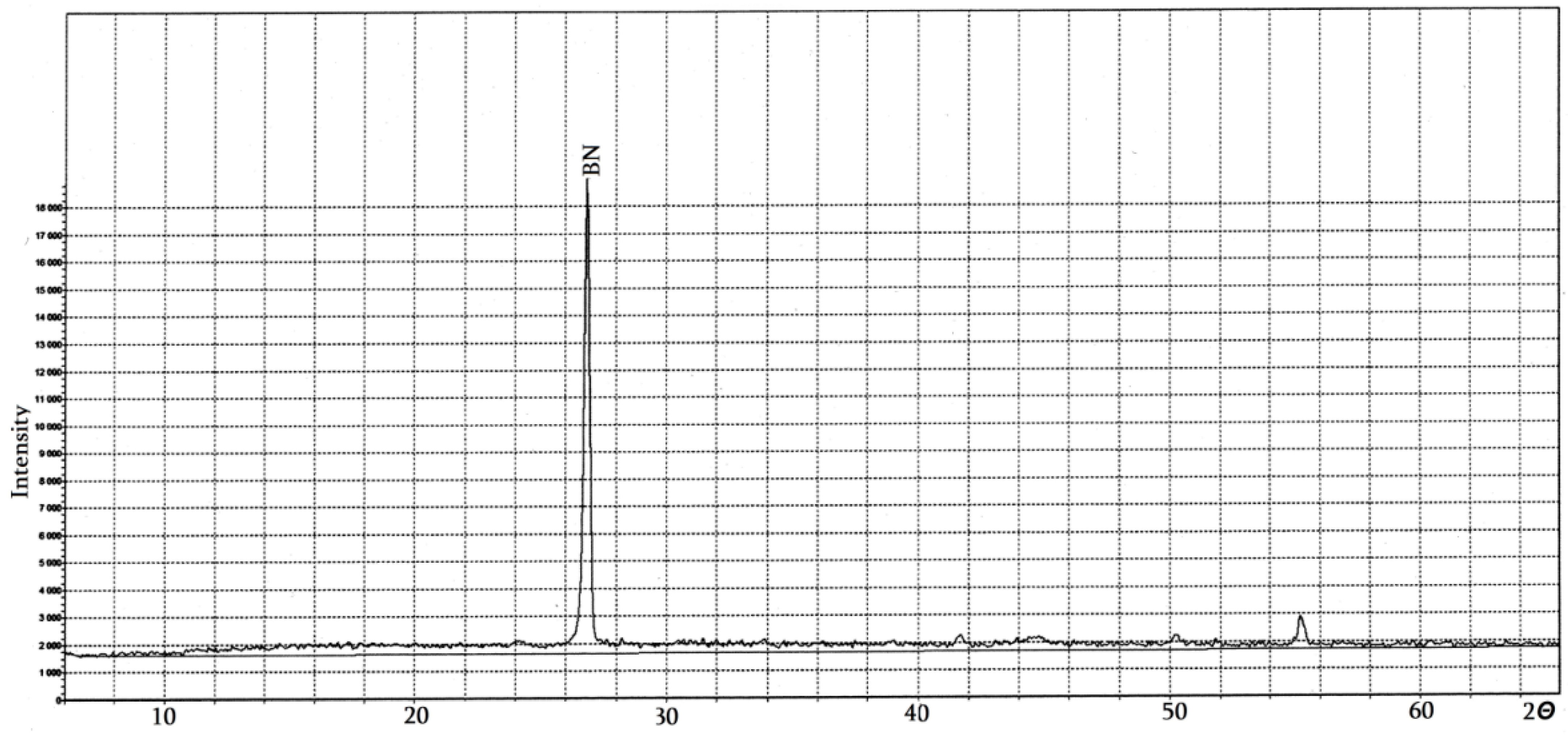
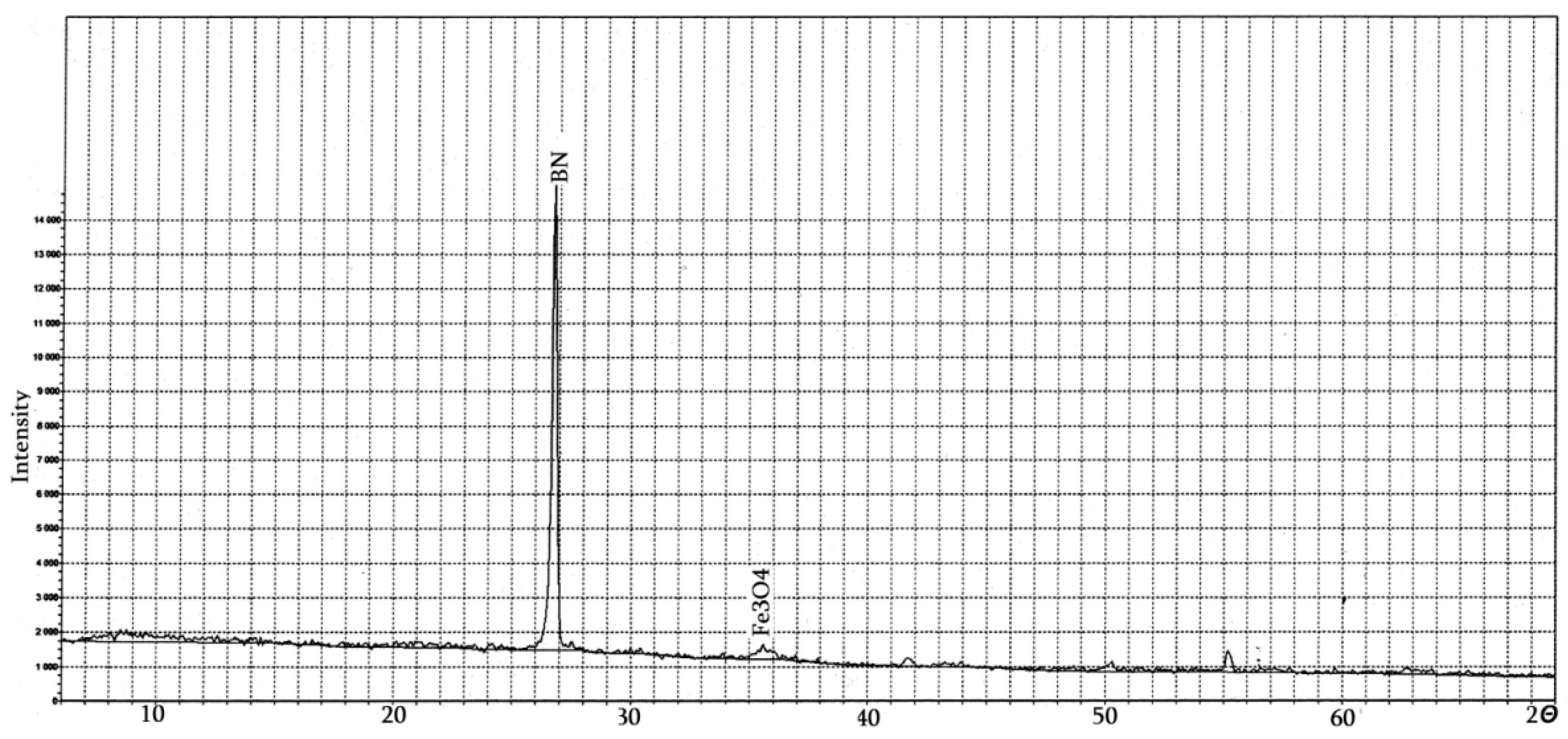
4.2. Pure Hexagonal Boron Nitride
4.3. Hexagonal Boron Nitride–Iron Composite
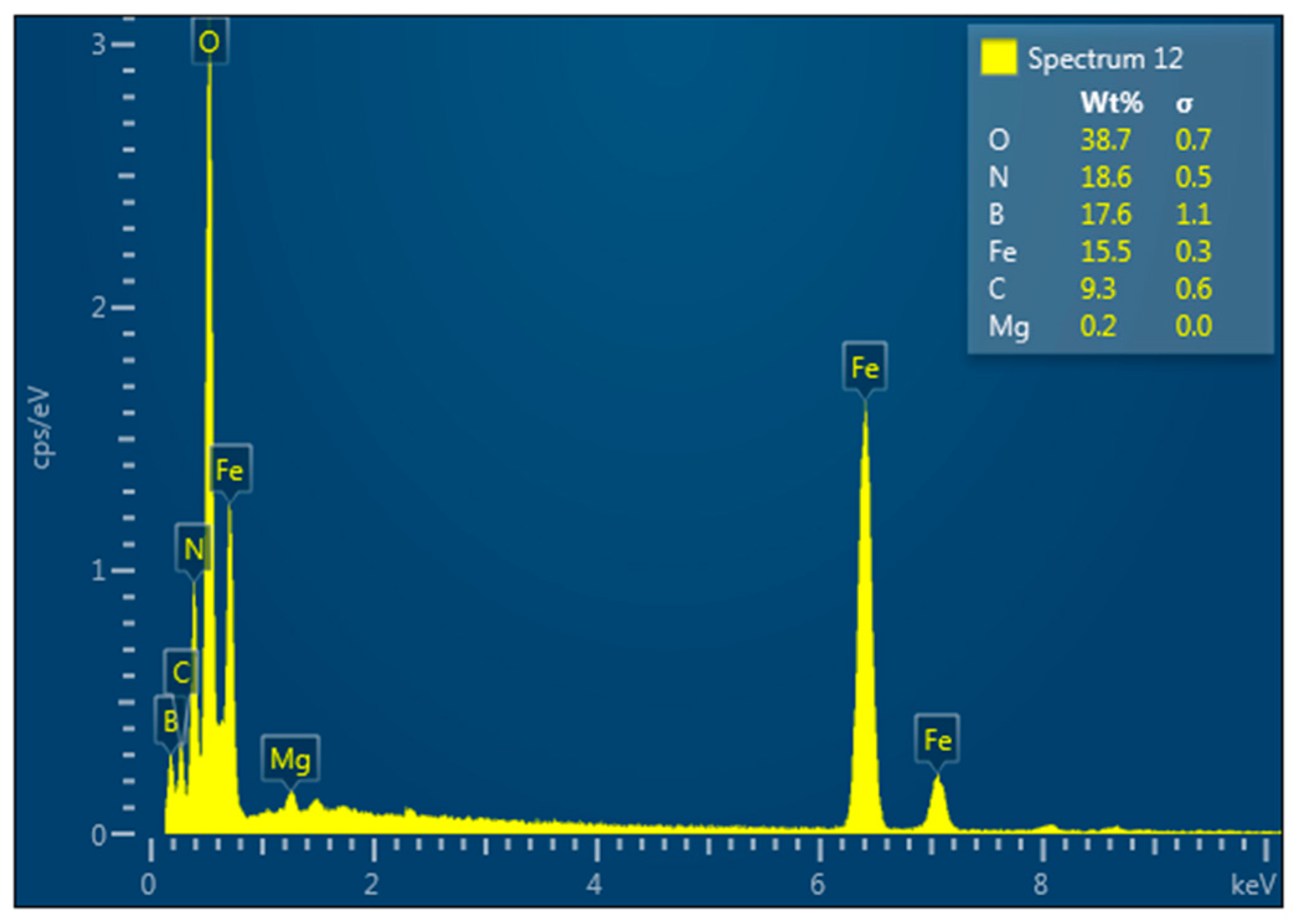
4.4. Hexagonal Boron Nitride–Magnetite Composite
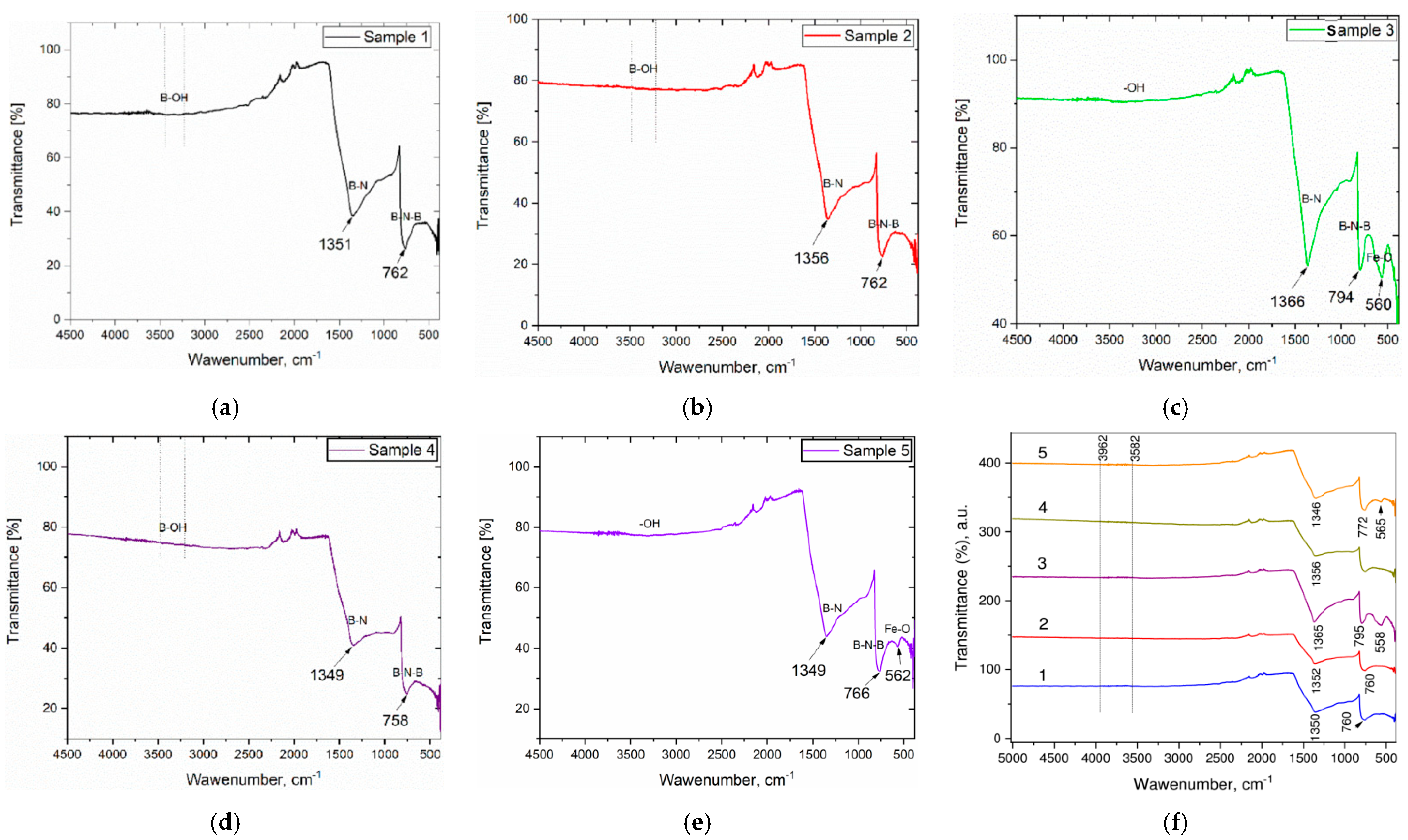
4.5. Fourier Transform Infrared Spectra
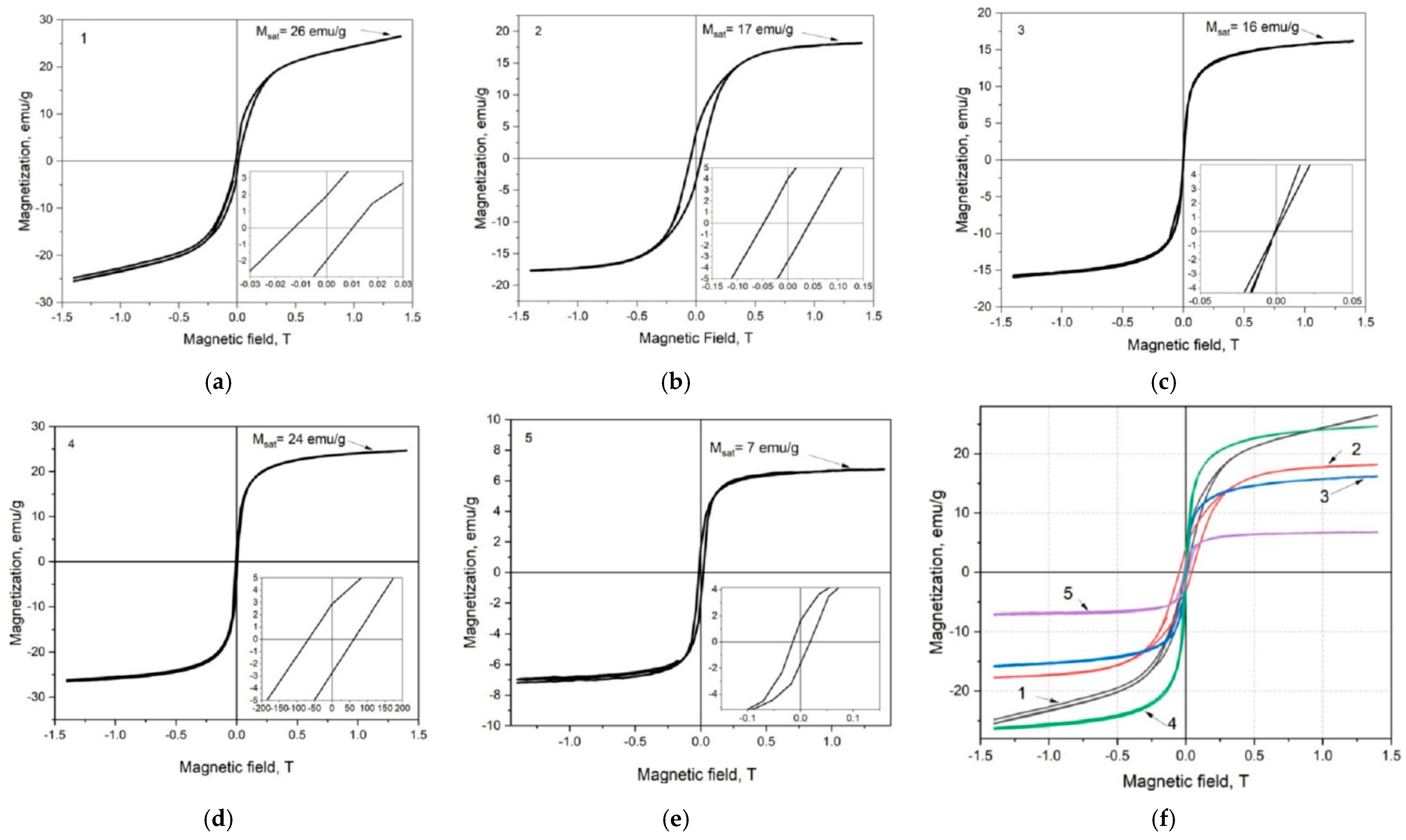
4.6. Magnetic Properties
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chkhartishvili, L.; Chedia, R.; Tsagareishvili, O.; Mirzayev, M.; Makatsaria, S.; Gogolidze, N.; Barbakadze, N.; Buzariashvili, M.; Lekashvili, O.; Jinikashvili, I. Preparation of neutron-capturing boron-containing nanosystems. In Proceedings of the 9th International Conference and Exhibition on Advanced and Nano Materials, Victoria, BC, Canada, 24–26 October 2022; IAEMM: Victoria, BC, Canada, 2022; pp. 1–15, ISBN 978-1-77835-171-6. Available online: https://iaemm.com/Pubdetails.php (accessed on 1 August 2023).
- Chkhartishvili, L.; Makatsaria, S.; Gogolidze, N. Boron-containing fine-dispersive composites for neutron-therapy and neutron-shielding. In Proceedings of the International Scientific-Practical Conference “Innovations and Modern Challenges—2022”, Tbilisi, Georgia, 18–19 November 2022; Publishing House “Technical University”: Tbilisi, Georgia, 2023; pp. 221–226, ISBN 978-9941-28-944-6. Available online: https://publishhouse.gtu.ge/en/ (accessed on 1 August 2023).
- Nabakhtiani, G.; Chkhartishvili, L.; Gigineishvili, A.; Tsagareishvili, O.; Gabunia, D.; Rostomashvili, Z.; Dekanosidze, S. Attenuation of gamma-radiation concomitant neutron-absorption in boron–tungsten composite shields. Nano Stud. 2013, 8, 259–266. Available online: https://www.nanostudies.org/index.php/nano/issue/archive (accessed on 1 August 2023).
- Evans, B.R.; Lian, J.; Ji, W. Evaluation of shielding performance for newly developed composite materials. Ann. Nucl. Energy 2018, 116, 1–9. [Google Scholar] [CrossRef]
- Chkhartishvili, L. Chapter 11. Boron-Containing Nanostructured Materials for Neutron-Shields. In Nanostructured Materials for the Detection of CBRN; Bonca, J., Kruchinin, S., Eds.; Springer Science: Dordrecht, The Netherlands, 2018; pp. 133–154. [Google Scholar] [CrossRef]
- Martin, P.M. Active thin films: Applications for graphene and related materials. Vac. Technol. Coat. 2018, 19, 6–14. Available online: https://digital.vtcmag.com/12727/10466/index.html?20400 (accessed on 1 August 2023).
- Zinovev, A.; Terentyev, D.; Chang, C.-C.; Yin, C.; Bakaev, A.; Rieth, M.; Lied, P.; Reiser, J.; Bonnekoh, C. Effect of neutron irradiation on ductility of tungsten foils developed for tungsten-copper laminates. Nucl. Mater. Energy 2022, 30, 101133. [Google Scholar] [CrossRef]
- Sorokin, O.; Kuznetsov, B.; Lunegova, Y.; Erasov, V. High-temperature composites with a multi-layered structure (Review). Proc. All-Russ. Sci. Res. Inst. Aviat. Mater. 2020, 88, 42–53. (In Russian) [Google Scholar] [CrossRef]
- Mann, K.S.; Mann, S.S. Py-MLBUF: Development of an online-platform for gamma-ray shielding calculations and investigations. Ann. Nucl. Energy 2021, 150, 107845. [Google Scholar] [CrossRef]
- Dai, M.; Zhang, Z.; Zhu, J.; Wang, X.; Xu, J.; Fu, X.; Bai, L.; Huang, Q.; Wang, Z.; Chen, L. Influence of interface roughness on reflectivity of tungsten/boron-carbide multilayers with variable bi-layer number by X-ray reflection and diffuse scattering. Chin. Opt. Lett. 2009, 7, 738–740. Available online: https://opg.optica.org/col/abstract.cfm?uri=col-7-8-738 (accessed on 1 August 2023).
- Makatsaria, S.; Chkhartishvili, L.; Dekanosidze, S.; Chedia, R. Nanopowder boron compounds doped with ferromagnetic clusters for BNCT. Int. J. Adv. Nano Comput. Anal. 2023, 2, 1–12. Available online: https://www.researchlakejournals.com/index.php/IJANCA/article/view/189 (accessed on 1 August 2023).
- Seneviratne, D.S.; Saifi, O.; Mackeyev, Y.; Malouff, T.; Krishnan, S. Next-generation boron drugs and rational translational studies driving the revival of BNCT. Cells 2023, 12, 1398. [Google Scholar] [CrossRef]
- Wang, Y.; Long, B.F.; Liu, C.Y.; Lin, G.A. Evolution of reduction process from tungsten oxide to ultrafine tungsten powder via hydrogen. High Temp. Mater. Process. 2021, 40, 171–177. [Google Scholar] [CrossRef]
- Barbakadze, N.; Sarajishvili, K.; Chedia, R.; Chkhartishvili, L.; Tsagareishvili, O.; Mikeladze, A.; Darchiashvili, M.; Ugrekhelidze, V. Obtaining ultrafine powders of some boron carbide-based nanocomposites using liquid precursors. Nanotechnol. Percept. 2019, 15, 243–256. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Mikeladze, A.; Chedia, R.; Tsagareishvili, O.; Barbakadze, N.; Sarajishvili, K.; Darchiashvili, M.; Ugrekhelidze, V.; Korkia, T. Synthesizing fine-grained powders of complex compositions B4C–TiB2–WC–Co. Solid State Sci. 2020, 108, 106439. [Google Scholar] [CrossRef]
- Barbakadze, N.; Chkhartishvili, L.; Mikeladze, A.; Tsagareishvili, O.; Sarajishvili, K.; Korkia, T.; Darchiashvili, M.; Rurua, L.; Jalabadze, N.; Chedia, R. Method of obtaining multicomponent fine-grained powders for boron carbide matrix ceramics production. Mater. Today Proc. 2022, 51, 1863–1871. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Mikeladze, A.; Jalabadze, N.; Nadaraia, L.; Korkia, T.; Chedia, R. New low-temperature method of synthesis of boron carbide matrix ceramics ultra-dispersive powders and their spark plasma sintering. Solid State Phenom. 2022, 331, 173–184. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Mikeladze, A.; Chedia, R.; Tsagareishvili, O.; Bugdayci, M.; Karagoz, I.; Maras, T.; Jalabadze, N.; Kvatchadze, V. Chapter 4: Combustion synthesis of boron carbide matrix for superhard nanocomposites production. In Advances in Combustion Synthesis and Technology; Bugdayci, M., Oncel, L., Eds.; Bentham Science Publisher: Singapore, 2022; pp. 66–95. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Mikeladze, A.; Tsagareishvili, O.; Kvatchadze, V.; Tavkhelidze, V.; Mestvirishvili, Z.; Driaev, D.; Barbakadze, N.; Nadaraia, L.; Sarajishvili, K.; et al. Advanced boron carbide matrix nanocomposites obtained from liquid-charge: Focused review. Condens. Matter 2023, 8, 37. [Google Scholar] [CrossRef]
- Hung, C.-C.; Hurst, J.; Santiago, D.; Rogers, R.B. Exfoliation of Hexagonal Boron Nitride via Ferric Chloride Intercalation (NASA/TM–2014-218125); NASA Glenn Research Center: Cleveland, OH, USA, 2014; pp. 1–20. Available online: https://ntrs.nasa.gov/api/citations/20140005373/downloads/20140005373.pdf (accessed on 1 August 2023).
- Píš, I.; Nappini, S.; Bondino, F.; Menteş, T.O.; Sala, A.; Locatelli, A.; Magnano, E. Fe intercalation under graphene and hexagonal boron nitride in-plane heterostructure on Pt(111). Carbon 2018, 134, 274–282. [Google Scholar] [CrossRef]
- Patel, R.B.; Liu, J.; Eng, J.; Iqbal, Z. One-step CVD synthesis of a boron nitride nanotube–iron composite. J. Mater. Res. 2011, 26, 1332–1339. [Google Scholar] [CrossRef]
- Chang, M.; Leung, C.; Wang, D.N.; Cheng, D. Process for CVD Deposition of Tungsten Layer on Semiconductor Wafer. US Patent 5028565, 2 July 1991. Available online: https://patentimages.storage.googleapis.com/f2/06/0f/1d36151bc50532/US5028565.pdf (accessed on 1 August 2023).
- Kim, S.H. Deposition of tungsten thin film on silicon surface by low pressure chemical vapor deposition method. J. Korean Chem. Soc. 1994, 38, 473–479. [Google Scholar]
- Plyushcheva, S.V.; Mikhailov, G.M.; Shabel’nikov, L.G.; Shapoval, S.Y. Tungsten thin-film deposition on a silicon wafer: The formation of silicides at W–Si interface. Inorg. Mater. 2009, 45, 140–144. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, J.-H.; Sohn, I.-H.; Hwang, T.-J.; Lee, K.-Y. Preparation of tungsten metal film by spin coating method. Korea–Aust. Rheol. J. 2002, 14, 71–76. Available online: https://www.cheric.org/PDF/KARJ/KR14/KR14-2-0071.pdf (accessed on 1 August 2023).
- Singla, G.; Singh, K.; Pandey, O. Structural and thermal properties of in-situ reduced WO3 to W powder. Powder Technol. 2013, 237, 9–13. [Google Scholar] [CrossRef]
- Yu, M.L.; Ahn, K.Y.; Joshi, R.V. Surface reactions in the chemical vapor deposition of tungsten using WF6 and SiH4 on Al, PtSi, and TiN. J. Appl. Phys. 1990, 67, 1055–1061. [Google Scholar] [CrossRef]
- Gao, J.; Chan, L.H.; Wongsenakhum, P. Methods for Improving Uniformity and Resistivity of Thin Tungsten Films. US Patent 7655567B1, 2 February 2010. Available online: https://patentimages.storage.googleapis.com/0d/c7/0b/6be43db1030817/US7655567.pdf (accessed on 1 August 2023).
- Yang, M.; Aarnink, A.A.I.; Kovalgin, A.Y.; Gravesteijn, D.J.; Wolters, R.A.M.; Schmitz, J. Comparison of tungsten films grown by CVD and hot-wire assisted atomic layer deposition in a cold-wall reactor. J. Vac. Sci. Technol. A 2016, 34, 01A129. [Google Scholar] [CrossRef]
- Dippel, A.-C.; Schneller, T.; Lehmann, W.; Reichenberg, B.; Waser, R. Tungsten coatings by chemical solution deposition for ceramic electrodes in fluorescent tubes. J. Mater. Chem. 2008, 18, 3501–3506. [Google Scholar] [CrossRef]
- Cao, P.; Cao, J.-P.; Cao, J.-H. Boron Carbide Ceramic Metallization Preparation Method. CN Patent CN110981550B, 7 December 2021. (In Chinese). Available online: https://eureka.patsnap.com/pdfnew/?patentId=cede242d-11a6-41e5-880a-e2a2dfb9ae88 (accessed on 1 August 2023).
- Mallia, B.; Dearnley, P. Exploring new W–B coating materials for the aqueous corrosion–wear protection of austenitic stainless steel. Thin Solid Films 2013, 549, 204–215. [Google Scholar] [CrossRef]
- Guldamashvili, A.; Nardaya, Y.; Nebieridze, T.; Sanaia, E.; Sichinava, A.; Kadaria, M. Mechanical properties of tungsten implanted with boron and carbon ions. J. Mater. Sci. Eng. A 2017, 7, 82–88. [Google Scholar] [CrossRef]
- Gromilov, S.A.; Kinelovskii, S.A.; Alekseev, A.V.; Kirienko, I.B. Investigation of W2B and β-WB high-temperature phases in coatings produced by a shaped charge explosion. J. Struct. Chem. 2010, 51, 1126–1131. [Google Scholar] [CrossRef]
- Szwacki, N.G. The structure and hardness of the highest boride of tungsten, a borophene-based compound. Sci. Rep. 2017, 7, 4082. [Google Scholar] [CrossRef]
- Mazo, I.; Molinari, A.; Sglavo, V.M. Electrical resistance flash sintering of tungsten carbide. Mater. Des. 2022, 213, 110330. [Google Scholar] [CrossRef]
- Syrovatko, Y.V. Calculation of the entropy of the eutectic phases WC and W2C in alloy W–C by the method of statistical processing of photo-images. Ukr. Appl. Phys. 2020, 4, 79–84. (In Russian). Available online: https://applphys.orion-ir.ru/appl-20/20-4/PF-20-4-79.pdf (accessed on 1 August 2023).
- Pankratz, L.B. Thermodynamic Properties of Carbides, Nitrides, and Other Selected Substances; U.S. Department of the Interior: Washington, DC, USA, 1994; Bulletin 696, pp. 900–901. Available online: https://digital.library.unt.edu/ark:/67531/metadc12836/ (accessed on 1 August 2023).
- Song, J.; Huang, C.; Zou, B.; Liu, H.; Liu, L.; Wang, J. Effects of sintering additives on microstructure and mechanical properties of TiB2–WC ceramic–metal composite tool materials. Int. J. Refract. Met. Hard Mater. 2012, 30, 91–95. [Google Scholar] [CrossRef]
- Zhang, C.; Song, J.; Jiang, L.; Gao, J.; Liang, G.; Lei, C.; Xie, J.; Wang, S.; Lv, M. Fabrication and tribological properties of WC–TiB2 composite cutting tool materials under dry sliding condition. Tribol. Int. 2017, 109, 97–103. [Google Scholar] [CrossRef]
- Ozer, S.C.; Buyuk, B.; Tugrul, A.B.; Turan, S.; Yucel, O.; Goller, G.; Sahin, F.C. Gamma and neutron shielding behavior of spark plasma sintered boron carbide–tungsten based composites. In TMS 2016 145th Annual Meeting Supplemental Proceedings, Nashville, TN, USA, 14–18 February 2016; The Minerals, Metals & Materials Society, Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 449–456. [Google Scholar] [CrossRef]
- Zhang, W.; Yamashita, S.; Kita, H. Progress in pressureless sintering of boron carbide ceramics—A review. Adv. Appl. Ceram. 2019, 118, 222–239. [Google Scholar] [CrossRef]
- Sugiyama, S.; Taimatsu, H. Preparation of WC–WB–W2B composites from B4C–W–WC powders and their mechanical properties. Mater. Trans. 2002, 43, 1197–1201. [Google Scholar] [CrossRef]
- Tamizifar, H.; Hadian, A.; Tamizifar, M. The Comparison between boron carbide (B4C) with other common inhibitors on physical and mechanical properties of WC/Co. Int. J. Mod. Phys. Conf. Ser. 2012, 5, 102–110. [Google Scholar] [CrossRef]
- Taran, A.V.; Garkusha, I.E.; Taran, V.S.; Muratov, R.M.; Skoblo, T.S.; Sidashenko, O.I.; Romaniuk, S.P.; Maltsev, T.V.; Baturin, A.A. Structure and properties of B4C coatings obtained by RF sputtering with external magnetic field. In Nanomaterials and Nanocomposites, Nanostructure Surfaces, and Their Applications; Fesenko, O., Yatsenko, L., Eds.; Springer Nature: Cham, Switzerland, 2021; pp. 51–57. [Google Scholar] [CrossRef]
- Ma, K.; Shi, X.; Cao, X.; Yang, Z.; Zuo, J.; Xu, J.; Li, M. Mechanical, electrical properties and microstructures of hot-pressed B4C–WB2 composites. Ceram. Int. 2022, 48, 20211–20219. [Google Scholar] [CrossRef]
- Silvestroni, L.; Failla, S.; Gilli, N.; Melandri, C.; Savacı, U.; Turan, S.; Sciti, D. Disclosing small scale length properties in core-shell structured B4C–TiB2 composites. Mater. Des. 2021, 197, 109204. [Google Scholar] [CrossRef]
- Hofmann, H.; Petzow, G. Structure and properties of reaction hot-pressed B4C–TiB2–W2B5 materials. J. Less-Common Met. 1986, 117, 121–127. [Google Scholar] [CrossRef]
- Cai, K.-F.; Nan, C.-W. The influence of W2B5 addition on microstructure and thermoelectric properties of B4C ceramic. Ceram. Int. 2000, 26, 523–527. [Google Scholar] [CrossRef]
- Yeh, C.; Wang, H. Preparation of tungsten borides by combustion synthesis involving borothermic reduction of WO3. Ceram. Int. 2011, 37, 2597–2601. [Google Scholar] [CrossRef]
- Yin, J.; Huang, Z.; Liu, X.; Zhang, Z.; Jiang, D. Microstructure, mechanical and thermal properties of in situ toughened boron carbide-based ceramic composites co-doped with tungsten carbide and pyrolytic carbon. J. Eur. Ceram. Soc. 2013, 33, 1647–1654. [Google Scholar] [CrossRef]
- Ozer, S.C.; Turan, S.; Sahin, F.C. Mechanical and microstructural properties of spark plasma sintered B4C−W2B5 composites. In Proceedings of the 18th International Metallurgy and Materials Congress, Istanbul, Turkey, 29 September−1 October 2016; UChTEA: Istanbul, Turkey, 2016; pp. 62–66. Available online: https://www.metalurji.org.tr/index.php/duyurular/529-immc-2016-bildiriler-kitabi-yayinlandi (accessed on 1 August 2023).
- Deng, J.; Zhou, J.; Feng, Y.; Ding, Z. Microstructure and mechanical properties of hot-pressed B4C/(W,Ti)C ceramic composites. Ceram. Int. 2002, 28, 425–430. [Google Scholar] [CrossRef]
- Shabalin, I.L. Ultra-High Temperature Materials IV: Refractory Carbides III (W Carbides). A Comprehensive Guide and Reference Book; Springer Nature: Cham, Switzerland, 2022; pp. i–xiv, 1–934. [Google Scholar] [CrossRef]
- Kostoglou, N.; Polychronopoulou, K.; Rebholz, C. Thermal and chemical stability of hexagonal boron nitride (h-BN) nanoplatelets. Vacuum 2015, 112, 42–45. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Shin, B.-G.; Lee, D.-E.; Yoon, K.-B. Preparation of PP/2D-nanosheet composites using MoS2/MgCl2- and BN/MgCl2-Bi supported Ziegler–Natta catalysts. Catalysts 2020, 10, 596. [Google Scholar] [CrossRef]
- Ortiz, D.G.; Pochat-Bohatier, C.; Cambedouzou, J.; Bechelany, M.; Miele, P. Exfoliation of hexagonal boron nitride (h-BN) in liquid phase by ion intercalation. Nanomaterials 2018, 8, 716. [Google Scholar] [CrossRef]
- Wang, J.; Ma, F.; Liang, W.; Wang, R.; Sun, M. Optical, photonic and optoelectronic properties of graphene, h-BN and their hybrid materials. Nanophotonics 2017, 6, 943–976. [Google Scholar] [CrossRef]
- Dee, G.; O’donoghue, O.; Rafferty, A.; Gannon, L.; McGuinness, C.; Gun’ko, Y.K. Boron nitride nanosheets functionalized with Fe3O4 and CoFe2O4 magnetic nanoparticles for nanofiltration applications. ACS Appl. Nano Mater. 2023, 6, 12526–12536. [Google Scholar] [CrossRef] [PubMed]
- Konopatsky, A.S.; Firestein, K.L.; Evdokimenko, N.D.; Kustov, A.L.; Baidyshev, V.S.; Chepkasov, I.V.; Popov, Z.I.; Matveev, A.T.; Shetinin, I.V.; Leybo, D.V.; et al. Microstructure and catalytic properties of Fe3O4/BN, Fe3O4(Pt)/BN, and FePt/BN heterogeneous nanomaterials in CO2 hydrogenation reaction: Experimental and theoretical insights. J. Catal. 2021, 402, 130–142. [Google Scholar] [CrossRef]
- Bangari, R.S.; Yadav, V.K.; Singh, J.K.; Sinha, N. Fe3O4-functionalized boron nitride nanosheets as novel adsorbents for removal of arsenic(III) from contaminated water. ACS Omega 2020, 5, 10301–10314. [Google Scholar] [CrossRef]
- Makatsaria, S.; Kekutia, S.; Markhulia, J.; Mikelashvili, V.; Chkhartishvili, L.; Chedia, R. Magnetic properties of nanopowder h-BN doped with Fe and Fe3O4 nanoclusters. Nano Stud. 2021–2022, 21/22, 287–292. [Google Scholar] [CrossRef]
- Lee, J.S.; Cha, J.M.; Yoon, H.Y.; Lee, J.-K.; Kim, Y.K. Magnetic multi-granule nanoclusters: A model system that exhibits universal size effect of magnetic coercivity. Sci. Rep. 2015, 5, 12135. [Google Scholar] [CrossRef]
- Binns, C. Chapter 1—Tutorial section on nanomagnetism. Front. Nanosci. 2014, 6, 1–32. [Google Scholar] [CrossRef]
- Avancini, T.G.; Souza, M.T.; de Oliveira, A.P.N.; Arcaro, S.; Alves, A.K. Magnetic properties of magnetite-based nano-glass-ceramics obtained from a Fe-rich scale and borosilicate glass wastes. Ceram. Int. 2019, 45, 4360–4367. [Google Scholar] [CrossRef]
- Darziyeva, T.A.; Alekperov, E.S.; Jabarov, S.H.; Mirzayev, M.N. Influence of heavy ions on the magnetic properties of Fe3O4 nanoparticles. Integr. Ferroelectr. Int. J. 2023, 232, 127–133. [Google Scholar] [CrossRef]
- Mirzayev, M. Personal Communication; Institute of Radiation Problems: Baku, Azerbaijan, 2023. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
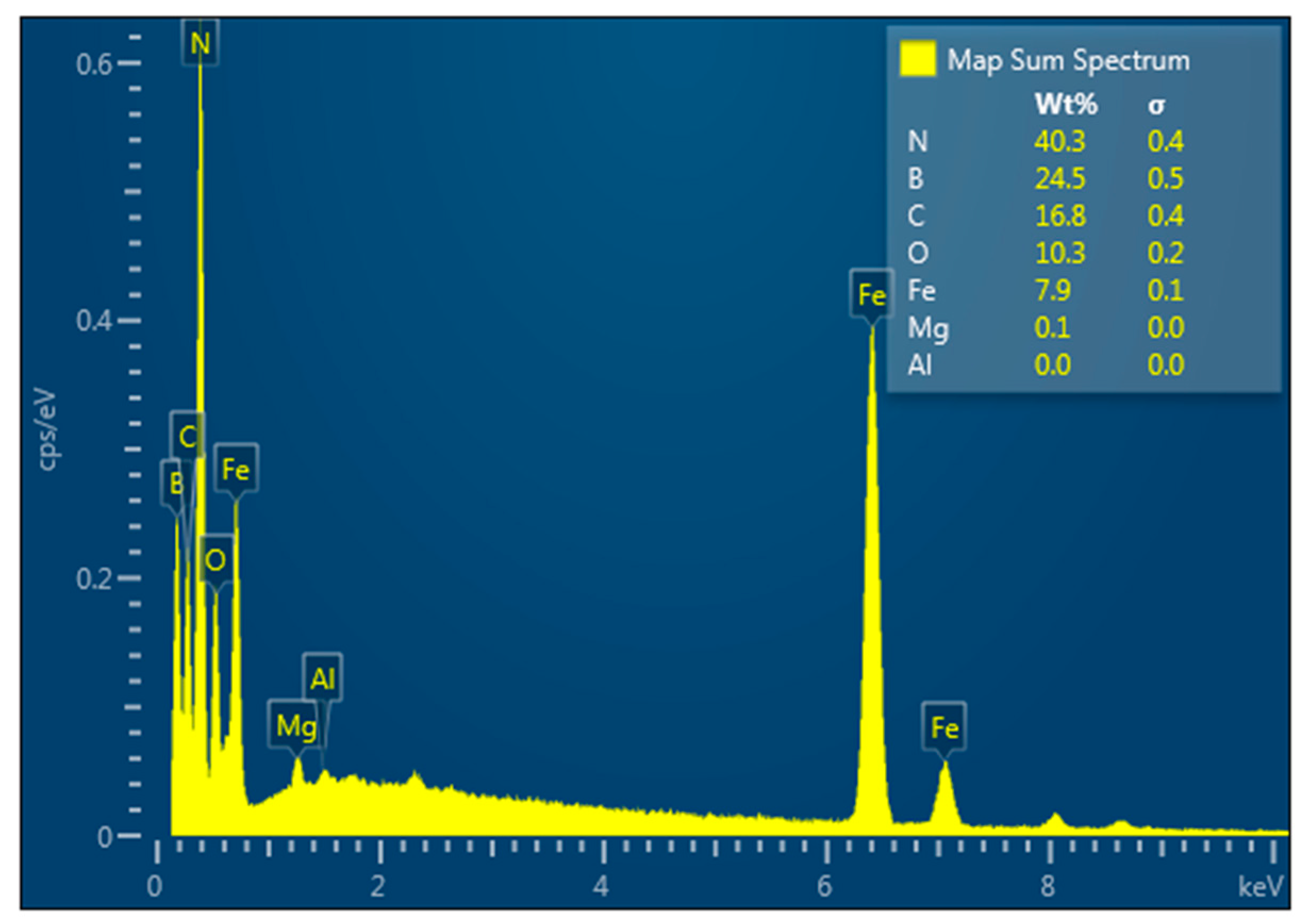
| Statistics | B | C | N | O | Mg | Al | Fe |
|---|---|---|---|---|---|---|---|
| Max | 24.52 | 16.79 | 40.34 | 10.28 | 0.11 | 0.04 | 7.91 |
| Min | 24.52 | 16.79 | 40.34 | 10.28 | 0.11 | 0.04 | 7.91 |
| Average | 24.52 | 16.79 | 40.34 | 10.28 | 0.11 | 0.04 | 7.91 |
| Deviation | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Spectrum Label | Content, wt.% |
|---|---|
| B | 24.52 |
| C | 16.79 |
| N | 40.34 |
| O | 10.28 |
| Mg | 0.11 |
| Al | 0.04 |
| Fe | 7.91 |
| Total | 100.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chkhartishvili, L.; Makatsaria, S.; Gogolidze, N.; Tsagareishvili, O.; Batsikadze, T.; Mirzayev, M.; Kekutia, S.; Mikelashvili, V.; Markhulia, J.; Minashvili, T.; et al. Obtaining Boron Carbide and Nitride Matrix Nanocomposites for Neutron-Shielding and Therapy Applications. Condens. Matter 2023, 8, 92. https://doi.org/10.3390/condmat8040092
Chkhartishvili L, Makatsaria S, Gogolidze N, Tsagareishvili O, Batsikadze T, Mirzayev M, Kekutia S, Mikelashvili V, Markhulia J, Minashvili T, et al. Obtaining Boron Carbide and Nitride Matrix Nanocomposites for Neutron-Shielding and Therapy Applications. Condensed Matter. 2023; 8(4):92. https://doi.org/10.3390/condmat8040092
Chicago/Turabian StyleChkhartishvili, Levan, Shio Makatsaria, Nika Gogolidze, Otar Tsagareishvili, Tamaz Batsikadze, Matlab Mirzayev, Shalva Kekutia, Vladimer Mikelashvili, Jano Markhulia, Tamaz Minashvili, and et al. 2023. "Obtaining Boron Carbide and Nitride Matrix Nanocomposites for Neutron-Shielding and Therapy Applications" Condensed Matter 8, no. 4: 92. https://doi.org/10.3390/condmat8040092