2.2.1. Irradiation
165Er has been the subject of some publications concerning various methods of production from a cyclotron or a neutron reactor. The neutron pathway leads to a very large range of activities but the result is “carrier added” (i.e., the presence of other isotopes of erbium). However, a ligand radiolabeling application has to be a “no carrier added” pathway. Therefore, irradiation with a cyclotron, starting from a different element, can be more interesting.
Irradiation of an erbium target (natural or enriched) by a proton beam will be according to the reactions:
natEr (p, xn)
165Tm → (by decay)
165Er or
natEr (d, xn)
165Tm →
165Er [
25]. The irradiation of this natural target will lead to
165Tm (30.06 h) but also to
167Tm (T ½ = 9.25 days) and
168Tm (T ½ = 93.1 days). Indeed, the percentage (%) of every isotope on natural erbium is 0.139, 1.601, 33.503, 22.869, 26.978 and 14.910 respectively to isotope 162, 164, 166, 167, 168 and 170 of erbium.
To overcome this, the irradiation of a 166Er enriched target guarantees a single nuclear reaction such as: 166Er (p, 2n) 165Tm which is quite challenging as it is not an easy and cheap way to obtain 165Er for proof of concept.
The “easiest” way to access to
165Er is the irradiation of a natural holmium target by proton beam according to the nuclear reaction
165Ho (p, n)
165Er at 16 MeV or deuteron beam by
165Ho (d, 2n)
165Er at 17.5 MeV. The proton beam method can be exploited in a commercial cyclotron (less than 20 MeV). At the cyclotron of Orléans, two ways of production have been explored. All cross sections were known and obtained by irradiation or simulation from calculation codes (ALICE-IPPE) [
12,
13,
26,
27].
The ratio 165Er/166Ho produced by proton and deuteron beam is respectively of around 400/1 and 8/1. The origin of the production of 166Ho by proton irradiation is not clearly identified, but neutron activation seems to be the source.
2.2.2. Separation 165Er/166Ho
The major difficulty in the development of
165Er is its extraction from the holmium target. The selectivity factor for the couple Ho/Er (SF: Separation Factor around 1.5) is amongst the lowest in the lanthanides. In all publications relative to the irradiation of a Ho target, only one evokes a treatment of this target by a separation of the Ho/Er [
11] pair. The complexing agent, α-HIBA (alpha-HydroxyIsoButyric Acid) is widely used in the separation of lanthanides [
28]. However, these separations are often made at the analytical level with the use of an online chromatographic system (High-Performance Liquid Chromatography (HPLC)) on balanced mixtures or traces (some ng or pg) for each lanthanide. To separate a few ng of
165Er from of few mg of a target of about 200 mg of natural holmium becomes more complex. In addition, the separation conditions must allow the use of
165Er in radiolabeling of ligands aiming vectorised therapy with Auger electron or used in radiotracer for biomodality MRI/SPECT.
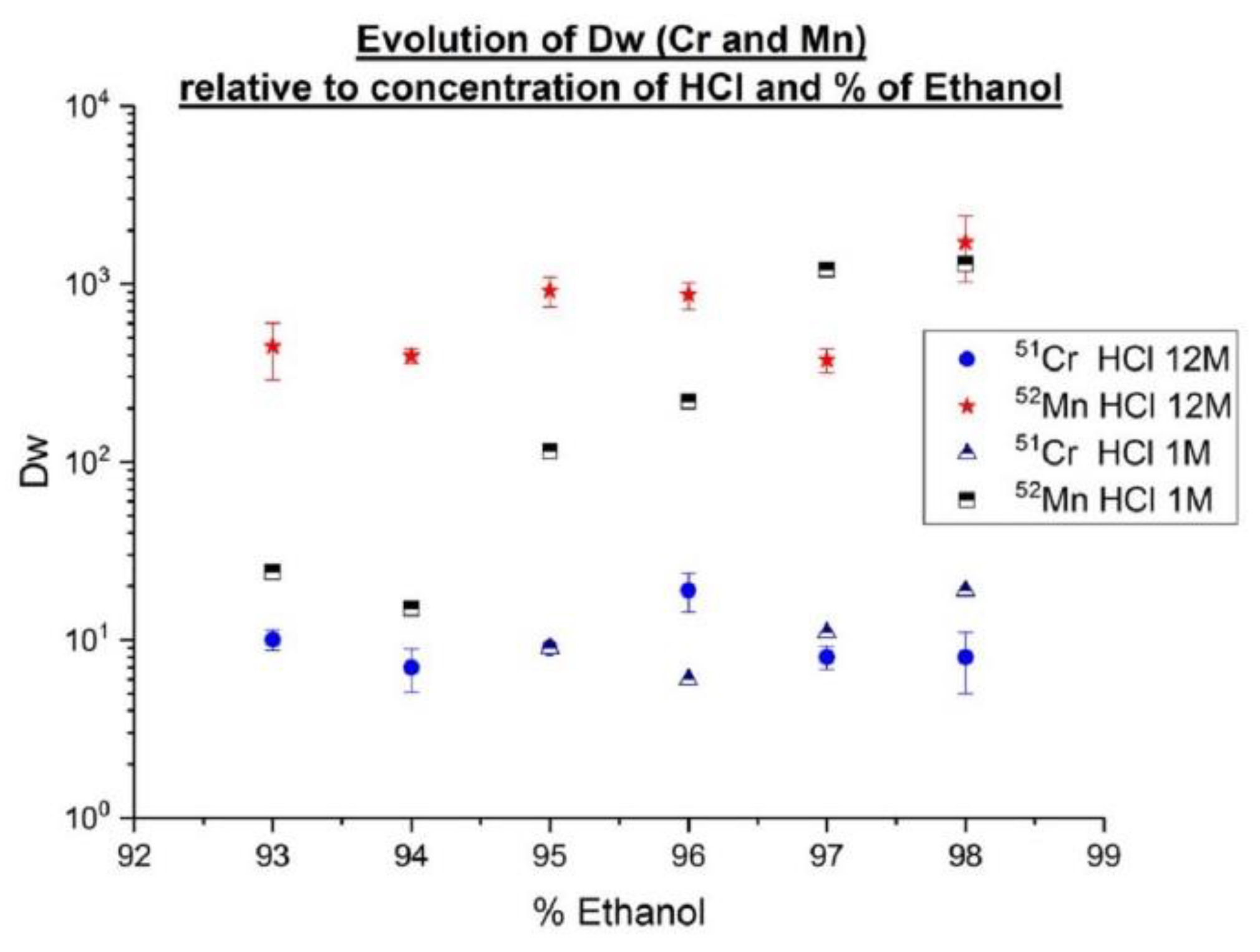
HDEHP (di(2-ethylhexyl) phosphoric acid), HEHEHP (mono-2-ethylhexyl ester of phosphonic acid) and (H[DTMPeP]) bis(2,4,4-trimethylpentyl) phosphinic acid are the main acidic organophosphorous extractants [
29] used in Rare Earth Element (REE) separation processes and have also been commercialized (for Triskem commercial name: LN, LN2 and LN3). HDEHP has been widely used for primary separation because the distribution coefficients of the REEs as a group differ markedly from impurities in leach groups (e.g., in spent fuel actinides and REEs) [
30]. For this project, LN2 resin (Triskem) based on (HEHEHP) was used. This reagent was chosen because REEs can be stripped at lower acidities than from HDEHP [
31]. They were used for the separation of adjacent lanthanides including the separation of a microcomponent from a macrocomponent [
32,
33,
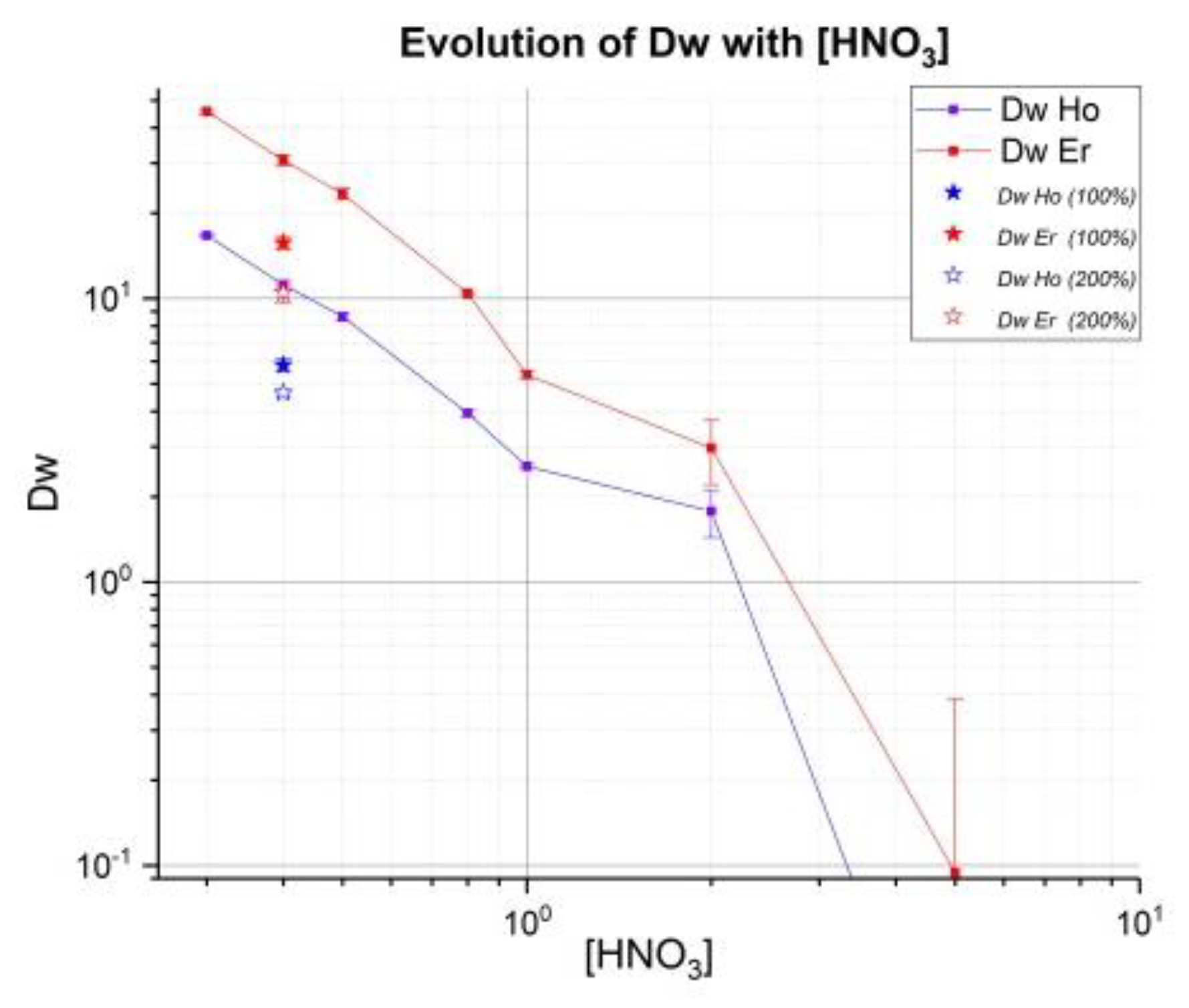
34]. Weight coefficients Dw have been determined for various concentrations of HNO
3. Additional evaluation of these coefficients on LN2 resin was performed with higher
166Ho activity using deuteron irradiation, which should allow more sensitivity. The results presented here are part of a more complete study about Ho/Er with comparison LN, LN2 and LN3 (in the case of LN3, it is necessary to use a low concentration of HNO
3). The main drawbacks of HDEHP (LN) is a low adjacent separation factor, relatively low acidity for REE stripping, as well as low hydrolytic stability [
35]. Phosphonic and phosphinic acids are more hydrolytically stable and effective extractants when compare with HDEHP. Moreover, they have demonstrated a high selectivity for the “heavy”REE (hREEs) compared to “light”REE (lREEs) [
29].
For determination of distribution coefficient Dw, a target of natural Ho of 0.3 mm of thickness (196 mg) was irradiated during 2 h at 2 µA at 17.5 Mev deuterons (AEOB: Activity (EOB)): around 700 MBq of 165Er/90 MBq of 166Ho were produced then dissolved in 1 mL of 5 M HNO3 solution (no cut target for batch tests) and after that the complete dissolution of the target, adjusted to 5.5 mL with 18 MΩ water. This solution of 0.9 M HNO3 was the crude solution (concentration around 35 mg/mL of holmium and 13 MBq of 165Er/1.6 MBq of 166Ho). Around 100 mg of LN2 resin was weighed for each test, which was always done in triplicate. In every tube, LN2 resin was mixed with 100 µL of crude solution and volume was adjusted to 1.5 mL with different volumes of HNO3 and water 18 MΩ to obtain the final concentration of HNO3.
For example, at 0.5 M HNO
3, 650 µL of 1 M HNO
3 and 750 µL of 18 MΩ water were added to 100 µL of crude solution of
165Er/
166Ho (initial concentration 0.9 M HNO
3). Every tube was shaken for 30 min [
36,
37] then allowed to rest and centrifuged for 20 min. 500 µL of each solution were extracted and diluted to 2 mL by adding 1.5 mL 18 MΩ water (geometry called as “Tube 2G”). Then samples were measured at spectrometry γ in this defined geometry with acceptable dead time (<5%). For several tubes, it was necessary to allow for one to two days of decay to perform a measurement.
Capacity of LN2 resin adsorption is 0.16 mmol/mL: maximum of 26.4 mg of holmium can be used for 0.37 g of resin (1 mL). However, Triskem recommends a surety factor of 50% on the capacity of adsorption. For that, in all batch tests 100 µL of crude solution contained around 3.5 mg of natural Ho. In a concentration of 0.4 M HNO3, two tests (denote 0.4 100 and 0.4 200) with 100% and 200% capacity of resin had added, respectively with 200 and 400 µL of crude solution to evaluate this factor on values Dw and SF.
Weight distribution D
w (mL/g) and separation factor (SF
Er/Ho) were calculated using the following equation:
Here A
0 and A
e are the liquid phase metal activities (Bq) before and after equilibrium,
m is the weight of the LN2 resin in grams and V is the volume of liquid phase in mL.
Then tests on a 2 mL column with around 0.37 g of LN2 resin (1 mL) were made first by the gravimetry method then with variation of flow (1 mL/min and 3 mL/min) at atmospheric pressure using a peristaltic pump in the outlet of the column. A gradient of concentration on HNO
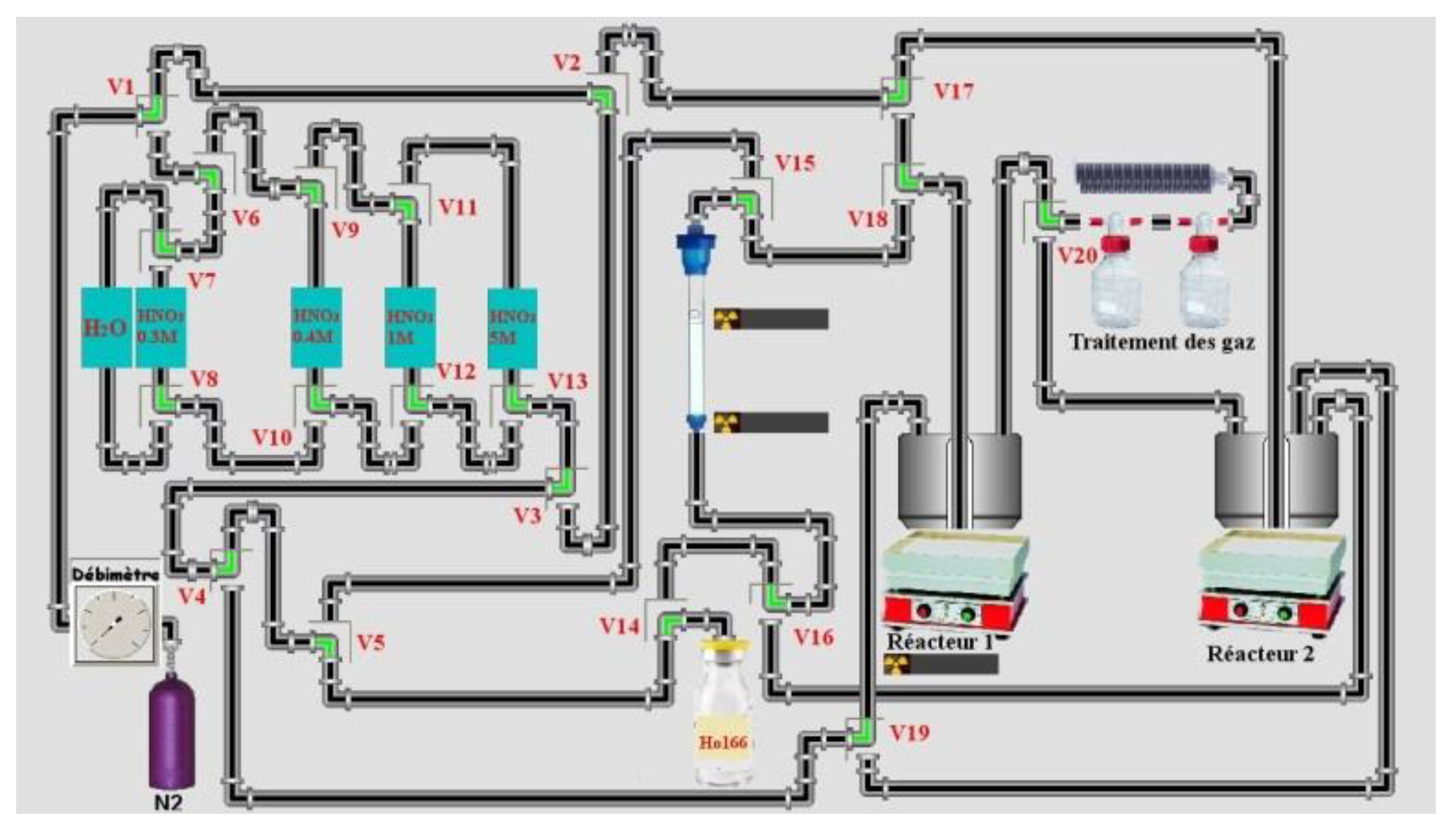
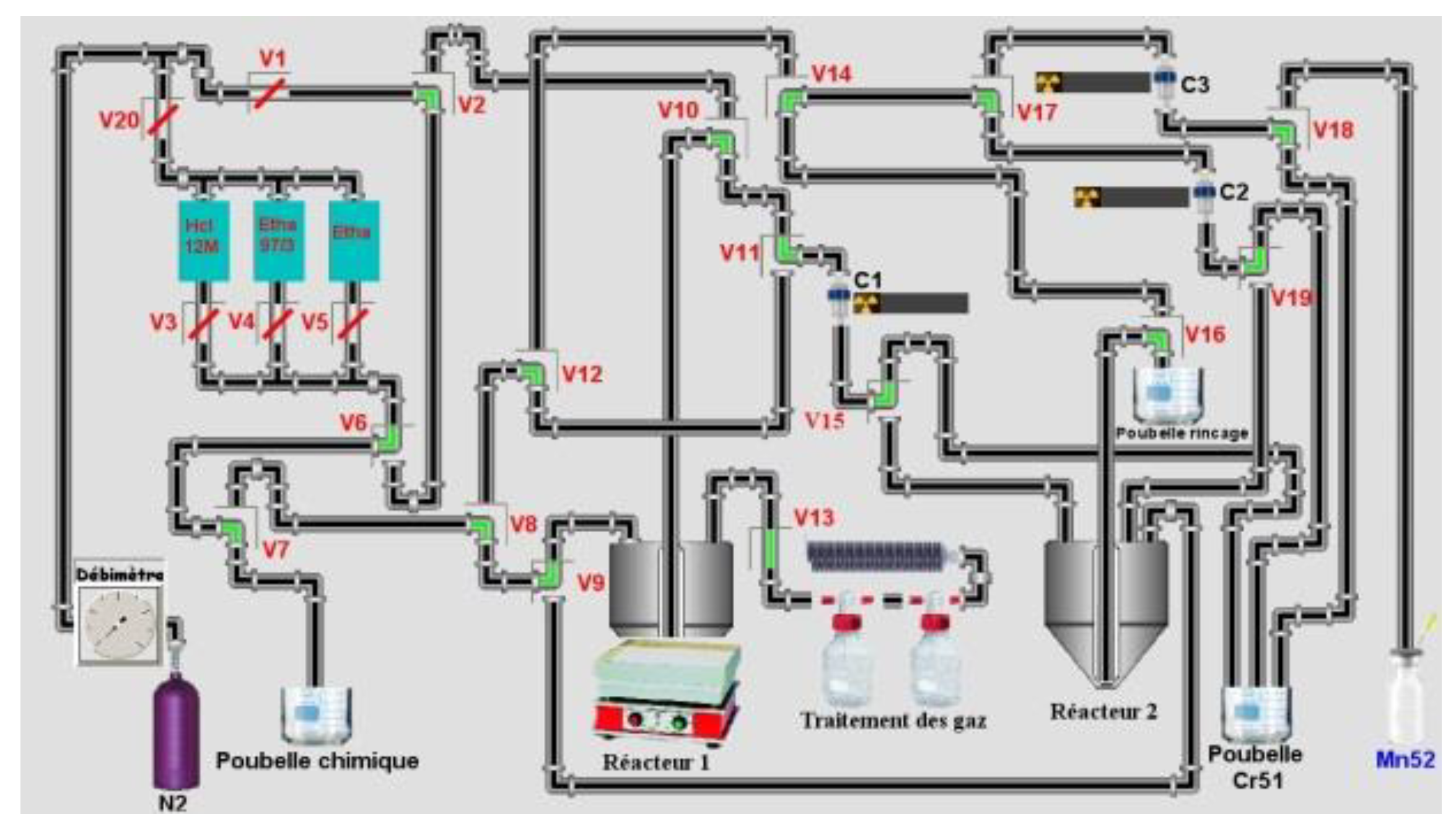
3 was used: 0.4 M 0.8 M, 1 M and 2 M or/and 5 M if necessary. Tests at 1 bar of Nitrogen (maximum pressure of solenoid valves from Bio-Chem (used in semi-automated system) is 1.4 bars) were made. Around 4 g of LN2 (for a maximum of mass target of 140 mg, around 11 mL) 50–90 mg of the target were used in semi-automated tests. Four tests on a homemade automation system (drive with the homemade software ACCRA (Automatisme & Contrôle Commande Radiochimie)) were made using the remote control sketch below (
Figure 3) from the dissolution step of the target until the final evaporation step of HNO
3.
In a semi-automated system 50 µL of solution were collected manually to determine the extraction yield of radionuclide in each test after adding 2 mL of 0.3 M HNO
3 in the reactor. The reactor was weighed before and after addition of 0.3 M HNO
3 (no weighing for test n°3: no extraction yield). This was the activity reference for
165Er and
166Ho (A
ref: deposited activity). With these values, the initial activity of
165Er and
166Ho in the cut target were determined. A relationship activity-weight was established by the ratio
165Ho/
166Ho: activity of
166Ho in Bq by mg of
165Ho (cut target). the initial ratio Ho/
165Er (for Ho,
165Ho was considered because the mass of
166Ho was insignificant) on the target (cut target mass) and the ratio in an isolated fraction of
165Er was calculated. A Ho decontamination factor was determined according to the following formula:
: Contamination factor in Ho
: Activity 166Ho in cut target
: Activity 166Ho in isolated fraction of 165Er
: Activity 165Er in cut target
: Activity 165Er in isolated fraction of 165Er
All dilution factors were determined by weighing aliquots of fraction. A geometry “Tube 2G” was used to determine the activities of the samples. Extraction yield (%) was defined as:
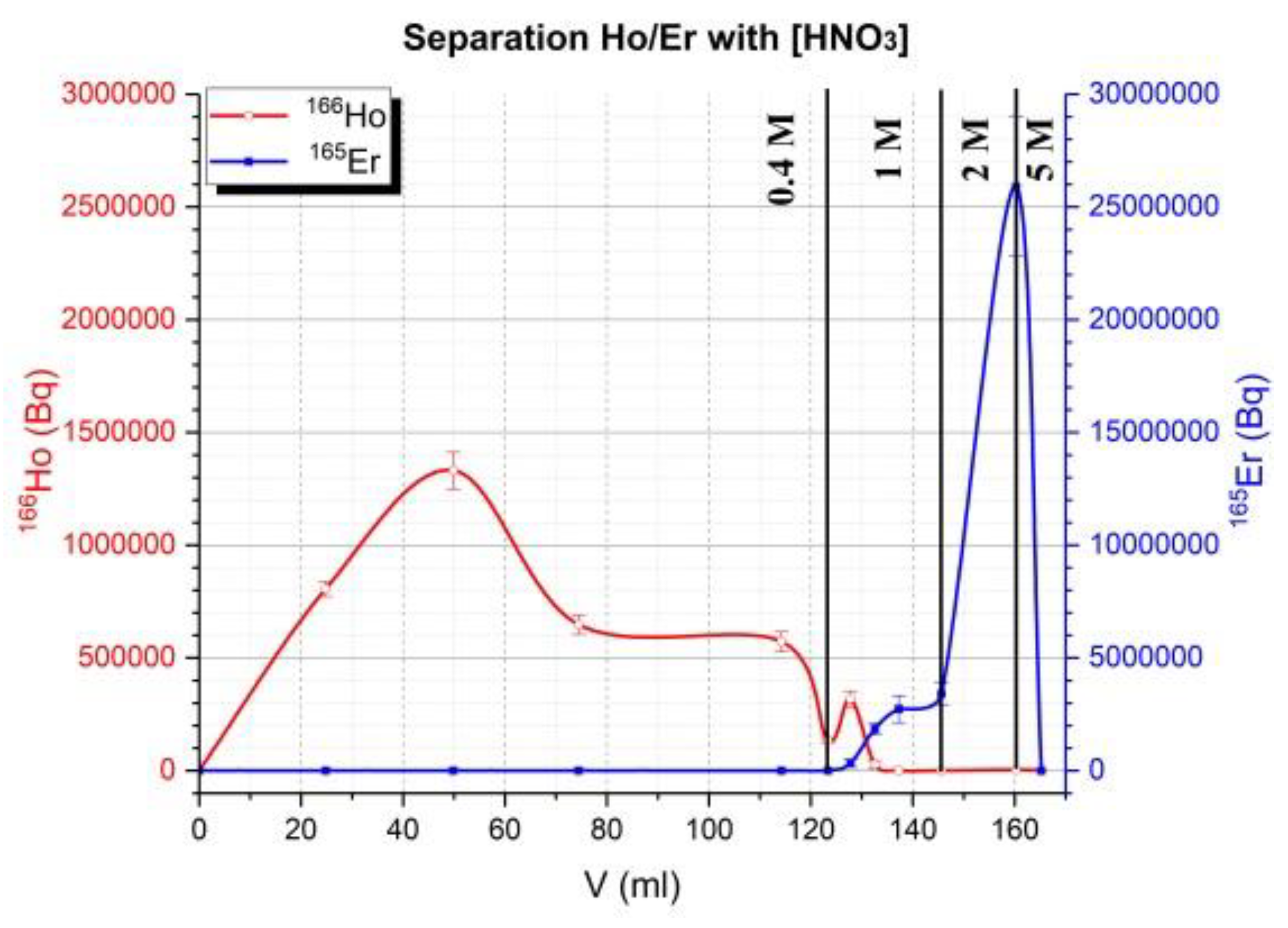
The same column was used and between different tests (4), column is conditioned with 30 mL of 5 M HNO3 then 18 MΩ water until pH was neutral. Then, 30 mL of 0.3 M HNO3 was used to conditioning column. Manually target was cut and around 50% of target was dissolved (more than 90% of activity of target). All weights listed for the target were referenced to cut the target (not all the irradiated mass of the target). The height of the column (glass) was 14 cm and the inner diameter 1 cm. The LN2 resin was shaken for 30 min with 18 MΩ water then put on the column. It was conditioned with 20 mL of 18 MΩ water then 30 mL of 0.3 M HNO3 before depositing the sample. Then, the cut target was dissolved in 1 mL of 5 M HNO3 in a reactor of 3 mL. Nitric acid was evaporated until it was in a pink gel formation. Then 2 mL of 0.3 M HNO3 was added to dissolve the pink residue and transfer it to the column under flow of nitrogen (20 mL/min). The solution on top of column was adsorbed under the flow of nitrogen (100 mL/min). When the solution on top of the column was adsorbed, 0.4 M HNO3 (100–120 mL) was used to elute Ho at flow of 1–2 drops/s (1 mL/min) under 1 bar of Nitrogen (at flow of 100 mL/min). Then 1 M HNO3 was used (around 40 mL except for sample 3 (22 mL)) and finally, in some cases, 2 M (15 mL for sample 3) and 5 M HNO3 (sample 1 and 3) more to elute residue activity on the column. The radiotracer method was used for this separation using 166Ho for Ho and 165Er for Er.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




