
In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer

1
Department of Chemistry, Drexel University, Philadelphia, PA 19104, USA
2
Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
*
Author to whom correspondence should be addressed.
Sci 2023, 5(4), 39; https://doi.org/10.3390/sci5040039
Submission received: 21 August 2023
/
Revised: 18 September 2023
/
Accepted: 30 September 2023
/
Published: 7 October 2023
(This article belongs to the Special Issue Feature Papers—Multidisciplinary Sciences 2023)
Abstract
:Context: Chronic inflammation has been linked to cancer since the 19th century. Tumor growth is supported by the proangiogenic factors that chronic inflammation requires. Polarized leukocytes initiate these angiogenic and tumorigenic factors. TIPE2, a transport protein, manages the cytoskeletal rearrangement that gives a polarized leukocyte its motility. Inhibition of this protein could lead to a therapeutic option for solid tumor cancers; however, no such inhibitors have been developed so far due to the large cavity size of the TIPE2 protein. Here we have examined possible small molecule inhibitors by combining structure-based and fragment-based drug design approaches. The highest binding ligands were complexed with the protein, and fragment libraries were docked with the complex with the intention of linking the hit compounds and fragments to design a more potent ligand. Three hit compounds were identified by in silico structure-based screening and a linked compound, C2–F14, of excellent binding affinity, was identified by linking fragments to the hit compounds. C2–F14 demonstrates good binding stability in molecular dynamic simulations and great predicted ADME properties. Methods: High throughput molecular docking calculations of mass libraries were performed using AutoDock Vina 1.1.2. Molecular docking of individual ligands was performed using AutoDock Vina with PyRx. Ligand libraries were prepared using OpenBabel, linked ligands were prepared using Avogadro. The protein was prepared using AutoDockTools-1.5.6. Protein-ligand complexes were visualized with PyMOL. Two- and three-dimensional representations of protein–ligand interactions were plotted with BIOVIA Discovery Studio Visualizer. In silico absorption, distribution, metabolism, and excretion (ADME) properties were calculated using SwissADME. Molecular dynamics simulations were conducted with GROMACS.
1. Introduction
Cancer is the second leading cause of death in the United States [1] and is expected to be the leading cause of death globally in the 21st century [2]. Tumor growth often arises at sites of chronic inflammation [3,4,5,6,7,8,9,10,11] as chronic inflammation establishes an environment that supports angiogenesis and tumorigenesis [4,7,11,12,13,14,15]. Cytokines, pro-inflammatory signaling proteins, are the link between inflammation and cancer [7]. Cytokines signal for the polarization of leukocytes, enabling the migration of leukocytes into inflamed areas, where leukocytes then initiate angiogenic and tumorigenic promoting factors. Tumor necrosis factor a-induced protein 8 like 2, or TIPE2, through its transportation of two phosphoinositides, controls the formation of both the leading and trailing edge of a polarized leukocyte. Inhibition of this protein would prevent the initial and secondary binding events that begin a sequence of actions that lead to the sustainment of chronic inflammation and subsequently, tumor cell proliferation, migration, and survival.
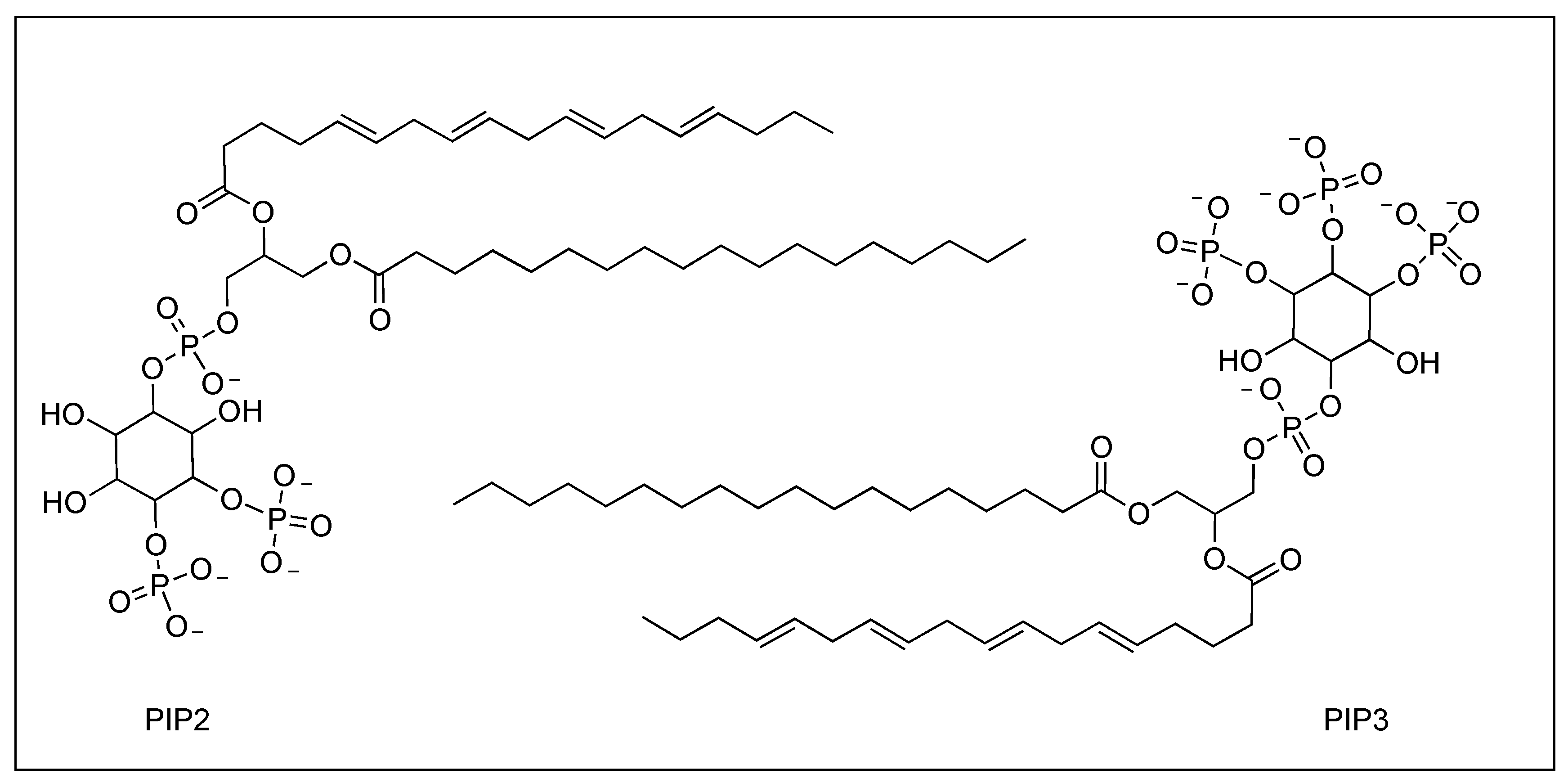
TIPE2 is a transport protein discovered by Sun et al. in the inflamed tissue of mice with experimental autoimmune encephalomyelitis (EAE) [16]. TIPE2, located on the human chromosome 1 and predominately expressed in lymphoid and myeloid cells, is essential in maintaining immune homeostasis as a regulator of innate and adaptive immune response [17,18]. Consisting of 150 amino acids, TIPE2 is a large protein with a fold distinctive to the tumor necrosis factor α-induced protein like 8 family. TIPE2 is characterized by a large central cavity of hydrophobic nature measuring 10 Å by 20 Å (Figure 1). This, in conjunction with its larger size, permits cofactors to fit in the cavity and bind to TIPE2 [18,19]. Through its role as a phosphoinositide transfer protein, TIPE2 can promote the formation of the leading edge of a polarized leukocyte. TIPE2 extracts phosphatidylinositol 4,5-bisphosphate (PIP2) from phosphatidylinositol 3,4,5-trisphosphate-rich (PIP3) membranes; this reduction in PIP2 levels promotes the formation of leading edges [13]. Once PIP2 (Figure 2) binds to TIPE2, TIPE2 subsequently transports and releases PIP2 to PI(3)K to be phosphorylated to PIP3. PIP3 then activates Rac-guanine-nucleotide-exchange factors which, in turn, activates Rac1 and further promotes the formation of the leading edge. TIPE2 plays dual roles in leukocyte polarization and can also bind to Rac1, inhibiting the latter’s function of promoting leading edges and, by default, enhancing the formation of the trailing edges.
Once the polarized leukocytes have reached the site of inflammation, they express several factors that promote angiogenesis: platelet-derived growth factor (PDGF), fibroblast growth factor-2 (FGF2), matrix metalloproteinase-9 (MMP-9), and vascular endothelial growth factor (VEGF) as well as tumor-promoting factor and tumor necrosis factor alpha (TNFα) [12,13], leading to the sustainment of an environment in which tumor cells can thrive.
TIPE2-deficient bone marrow neutrophils (BMNs), types of leukocytes, show a substantial decrease in polarization compared to wild-type BMNs. TIPE2-deficient mice with experimental autoimmune encephalomyelitis also exhibited diminished inflammation due to the reduction in leukocyte polarization and migration [18]. TIPE2 deficiency in myeloid-derived suppressor cells (MDSCs), a type of leukocyte that suppresses immune response, leads to reduced tumor growth [19]. These works by Fayngerts et al. and Yan et al. [18,19] indicate a therapeutic benefit for TIPE2 inhibition. However, no such inhibitors have been developed so far due to the large cavity size of the TIPE2 protein.
We have conducted a structure-based in silico screening, incorporating fragment-based design techniques, to identify potential TIPE2 inhibitors using AutoDock Vina. In the first step, three hit compounds from the two drug-like libraries of Enamine (consisting of around 200,000 molecules) and ChemBridge (consisting of around 350,000 molecules) were used. Then, to improve upon the binding affinity of the three hit compounds, three ligand–protein complexes were made, and then fragments were docked with the complexes. The fragment libraries were docked with the complex with the intention of linking the hit compounds and fragments to design a more potent ligand. Herein we present the results of the structure-based screening, supplying us with three hit compounds, and the highest binding hit-compound–fragment linked ligand.
2. Methods
High throughput molecular docking calculations of mass libraries were performed using AutoDock Vina 1.1.2 [20] on Drexel University’s computer cluster Proteus initially, which was replaced by Picotte. Molecular docking of individual ligands was performed using AutoDock Vina with PyRx [21] Ligand libraries were prepared using OpenBabel [22]; linked ligands were prepared using Schrodinger® docking suits (Schrödinger Maestro, New York, NY, USA. Version 11.9.011, MMshare Version 4.5.011) [23]. The protein was prepared using AutoDockTools-1.5.6 [24] Protein–ligand complexes were visualized with PyMOL [25]. Two- and three-dimensional representations of protein–ligand interactions were plotted with BIOVIA Discovery Studio Visualizer [26]. Molecular drawing was conducted with ChemDraw. In silico absorption, distribution, metabolism, and excretion (ADME) properties were calculated using SwissADME [27]. Molecular dynamics simulations were conducted with GROMACS [28].
2.1. Preparation of Receptor and Ligands
The crystal structure of TIPE2 was retrieved from the RCSB Protein Data Bank (PDB ID: 3F4M). The structure was cleaned, removing all water and co-crystallized atoms, and prepared with the addition of polar hydrogens and calculation of Gasteiger charges in AutoDockTools. Two “drug-like” libraries of ligands—Enamine, consisting of around 200,000 molecules, and ChemBridge (sourced from ZINC), consisting of around 350,000 molecules—were retrieved from their respective sites and optimized. Two fragment libraries—LC Advanced Subset of General Fragment Library and LC General Fragment Library Main Collection, consisting of around 70,000 compounds—were retrieved from the Life Chemicals website. Fragment libraries were also retrieved from Enamine, including a “mini-fragment” library, consisting of molecules around 100 amu. All libraries were prepared in OpenBabel through the addition of partial charges, minimization using the MMFF94, the Merck Molecular Force Field, and conversion to pdbqt format.
2.2. Preparation of Receptor and Hit Compound Complex
The protein and hit compounds were visualized together in PyMol and saved as one macromolecule for each protein–hit compound complex. The new macromolecule was imported into AutoDockTools, where it was prepared as a protein file in the aforementioned manner.
2.3. Molecular Docking
To inhibit the interaction of TIPE2 and PIP2, the grid box was initially constructed to cover the entire binding cavity of TIPE2. The grid box was centered at x = 63.57, y = 12.59, and z = 8.49, and of dimensions 25.00 Å × 25.00 Å × 25.00 Å along the x-, y-, and z-axis, respectively. This grid box was used to run the Enamine and ChemBridge “drug-like” libraries. The box was chosen to cover the entirety of the binding pocket; it is smaller than the protein but does capture the entire binding site. The fragment libraries were docked with a grid box that spans the binding cavity entrance, occluding the pocket, with dimensions of 25.00 Å × 25.00 Å × 20.00Å centered at x = 65.27, y = 12.22, z = 19.52. Top fragments were selected from the data based on binding affinity, the highest binding affinity being the most negative.
2.4. Fragment Modifications
Fragments were visualized in PyMol with the hit compound–protein complexes. The fragments were then linked to the hit compound using ChemDraw 8.0. The new compound was then minimized in Avogadro with the MMFF94 force field and then input into PyRx to be docked with the un-complexed protein in AutoDock Vina using the grid box of dimensions 25.00 Å × 25.00 Å × 20.00Å along the x-, y-, and z-axis, respectively, and centered at x = 65.27, y = 12.22, z = 19.52.
2.5. Molecular Dynamics
The three hit compound–protein complexes (C1–TIPE2, C2–TIPE2, and C3–TIPE2), the highest binding hit compound + fragment-protein complex (C2_F14–TIPE2) were solvated in a dodecahedron cell unit with the protein centered and at least 10 Å from the unit cell edge. The systems were neutralized with the appropriate Cl- or Na+ ion. Energy minimization was performed using the AMBERFF99SB-ILDN force field. Temperature was maintained at 300 K. MD simulations were run for 10 ns. Root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) were analyzed to examine the complexes’ binding stabilities.
3. Results and Discussion
3.1. Molecular Docking
Three hit compounds, (7S,9aR)-7-((1H-imidazol-4-yl)methyl)-2-((1-(p-tolyl)-1H-pyrazol-4-yl)methyl)hexahydro-2H-pyrazino [1,2-a]pyrazine-6,9-dione (compound 1, referred to herein as C1), 4-(3-((1,3a,4,6,7,7a-hexahydro-5H-pyrazolo [4,3-c]pyridin-5-yl)methyl)naphthalen-2-yl)piperazin-2-one (compound 2, referred to herein as C2), and (6-hydroxy-3a-methoxyoctahydro-1H-indol-1-yl)(2′-methyl-[1,1′-biphenyl]-3-yl)methanone (compound 3, referred to herein as C3) (Figure 3), were identified from the drug-like libraries for their high binding affinity. In addition to their binding affinity, C2 and C3 were chosen due to their higher binding position in the cavity. Docking scores of the compounds are compiled in Table 1.
C1 has a binding affinity of −8.7 kcalmol−1. Interactions between C1 and the protein are shown in Figure 4. As the compound sits lower in the cavity, interacting predominately with the hydrophobic amino acid residues that comprise the binding pocket, a lot of van der Waals interactions are exhibited. Phenylalanine 147, leucine 94, leucine 156, serine 151, leucine 155, leucine 159, isoleucine 171, leucine 175, leucine 120, alanine 98, leucine 116, alanine 101, glycine 97, valine 146, leucine 63, leucine 127, and valine 66 are involved in the hydrophobic interactions. A hydrogen bond is formed with phenylalanine 150 of length 2.19 Å. C1 interacts with the aliphatic residues alanine 67, valine 70, and leucine 43 by means of alkyl–alkyl interactions. Alanine 67 also participates in pi–alkyl interactions with the toluene moiety while phenylalanine 90 interacts with this group through pi–pi stacking.
C2 has a binding affinity of −8.8 kcalmol−1. N2 of the pyrazole (Figure 5) hydrogen bonds to serine 35 was found at the more hydrophilic entrance of the binding pocket with a bond length of 2.72 Å. The ligand is involved in van der Waals interactions with valine 70, valine 66, phenylalanine 147, leucine 178, leucine 63, valine 38, leucine 42, leucine 39, isoleucine 31, serine 36, arginine 91, leucine 184, and lysine 183. Phenylalanine 90 is involved in a T−shaped pi−pi interaction with the naphthalene moiety while alanine 67, leucine 127, and leucine 94 have pi−alkyl interactions.
C3 has a binding affinity of −8.7 kcalmol−1. Histidine 74 has a carbon−hydrogen bond with the methyl carbon of the ether (Figure 6). Van der Waals interactions are made with alanine 71 and leucine 94. Phenylalanine 90 and phenylalanine 147 exhibit pi−pi stacking, with phenylalanine 147 stacking in a T−shaped manner. Leucine 63 interacts with the toluene moiety in two ways, pi−alkyl interactions with the benzene ring and alkyl interactions with the methyl group. Valine 66, arginine 91, and valine 146 also exhibit alkyl−alkyl interactions. Alanine 67, valine 70, and leucine 127 interact with the second benzene ring through pi−alkyl interactions.
To improve upon the binding affinity of the three hit compounds, a fragment-based approach was utilized; three ligand−protein complexes were made, and then fragments were docked with the complexes. Due to the possible interactions with the complexed ligand and not only the protein, the binding affinities of the docked fragments are inflated. The top fragments (Figure 7) were visualized with the hit compounds and linked together, then docked with the un-complexed protein. Table 2 compiles the binding affinity of the hit compound–fragment linked ligands. A supplemental file has been added to show the structures of these compounds in Table 2.
The top section of Table 2 compiles the binding affinity of C1 linked to the top ten fragments F1–F10. Compound C1–F7 is the highest binding at −10.9 kcalmol−1. As depicted in Figure 8, two hydrogen bonds are established between serine 35 and both nitrogens cyclopentapyrazole of lengths 3.30 Å and 2.59 Å. A third hydrogen bond is formed between lysine 183 and the primary amine hydrogen of the piperazine dione. Lysine 183 also demonstrates pi−alkyl interaction with the methyl indene. Hydrophobic contacts form with residues alanine 67, leucine 94, leucine 184, alanine 71, aspartic acid 32, histidine 74, glycine 182, arginine 91, valine 70, phenylalanine 90, valine 66, and leucine 127. Phenylalanine 147 pi−pi stacks with the toluene moiety. Phenylalanine also interacts by means of alkyl−alkyl interactions with the methyl group of the toluene along with valine 146 and leucine 63. Leucine 63 also pi−alkyl interacts with the benzene of the toluene. Valine 38 and leucine 39 interact with the cyclopentapyrazole.
The highest binding linked ligand was C2–F14 or 2-(5-(7-((1,3a,4,6,7,7a-hexahydro-5H-pyrazolo [4,3-c]pyridin-5-yl)methyl)-6-(3-oxopiperazin-1-yl)naphthalen-1-yl)-2-oxo-2H-chromen-3-yl)quinazolin-4(3H)-one, with a binding affinity of −12.7 kcalmol−1; this is a significant improvement from the binding affinity of −8.7 kcalmol−1 of C2. The compound is predicted to exhibit poor aqueous solubility at a logS of −6.73; however, it has a decent partition coefficient of 2.87 MlogP and a topological polar surface area (TPSA) of 135.93 Å2. Two hydrogen bonds are formed between serine 35 and both nitrogens of the pyrazole of lengths 2.70 Å and 2.33 Å (Figure 9). The binding orientation of C2–F14 allows for the second hydrogen bond interaction that we do not see in the protein–ligand interactions of C2. Hydrophobic interactions are formed with leucine 184, aspartic acid 32, lysine 183, leucine 178, leucine 42, valine 28, leucine 39, isoleucine 68, phenylalanine 147, valine 146, valine 66, leucine 127, arginine 91, and histidine 74. Phenylalanine 90 pi−pi stacks with the center naphthalene moiety. Pi–alkyl interactions are exhibited by leucine 63, isoleucine 64, isoleucine 31, and leucine 94 with one of the four aromatic groups. Leucine 63 and valine 70 interact with the piperazinone. The type of interactions between TIPE2 and C2–14 are comparable to that of the interactions with C2; there are simply more interactions between TIPE2 and C2–14.
Structures of C3 linked to fragments F21−F30 are shown in the bottom section of Table 2. The highest binding affinity is given by C3−F25 with a score of −11.3 kcalmol−1. This ligand (Figure 10) exhibits van der Waals interactions with residues isoleucine 68, serine 35, leucine 63, leucine 184, leucine 94, valine 70, valine 66, leucine 127, phenylalanine 147, valine 146, valine 38, isoleucine 31, and aspartic acid 32. Pi−alkyl interactions are demonstrated by leucine 39 and alanine 67. Alkyl−alkyl interactions are exhibited by alanine 71, leucine 178, alanine 67, lysine 183, and arginine 91.
3.2. Molecular Dynamics
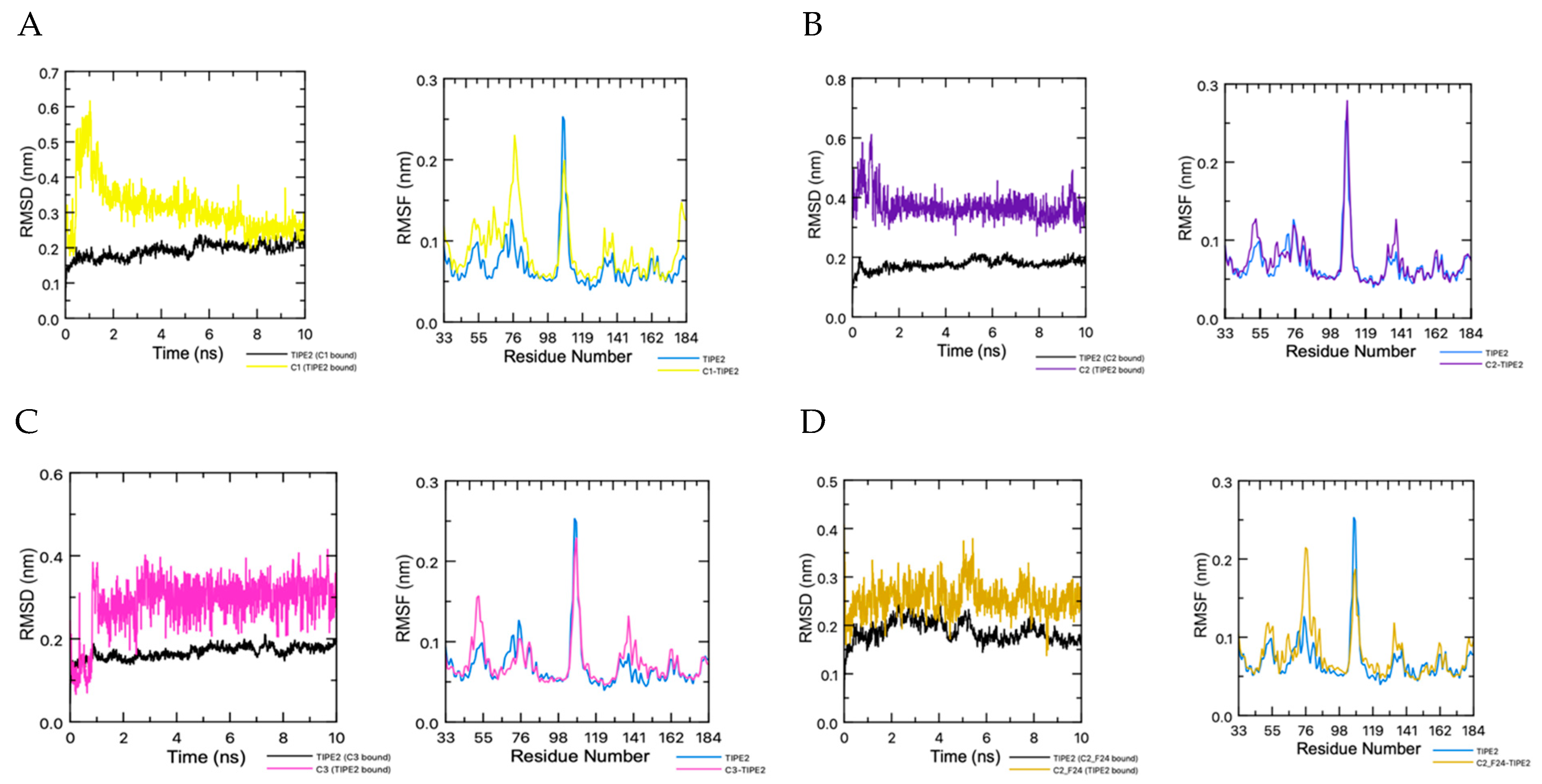
The binding complexes of the three hit compounds, C1–C3, and the top linked ligand, C2–F14, were investigated through MD studies. The RMSD plot in Figure 11A shows the initial instability of C1 in the binding complex. Within the first nanosecond, the ligand spikes from 0.2 nm to 0.6 nm. Within the next nanosecond, the ligand decreases to 0.35 nm and then continues to fall for the remainder of the simulation to around 0.2 nm, while fluctuating by 0.15 nm. The protein stabilizes around 6 ns. The RMSF plot depicts the significant fluctuations of the amino acid residues of the protein in the binding complex in comparison to the un-complexed ligand. The increased time in stabilization for this binding complex can be attributed to the position within the pocket of the ligand binding. C1 binds too far into the pocket whereas the rest of the ligands bind closer to the cavity entrance, confirming the validity of the grid box shift to higher in the cavity. The conformation of C1–F7 in the binding pocket fluctuates from 0.2 nm to 0.4 nm until it relaxes at 4 ns. Deviations are also seen in the protein with fluctuations from 0.2 nm to 0.3 nm until 4 ns. After 4 ns, the binding complex stabilizes with the ligand fluctuating at 0.30 nm and the protein at 0.2 nm. RMSD analysis of C2 depicts stabilization of the binding complex around 2 ns of the 10 ns MD simulation. After 2 ns, the ligand continually fluctuates at 0.375 nm. The ligand fluctuations increase around 9 ns to 0.50 nm before relaxing back to 0.375 nm (Figure 11B). The protein exhibits no deviations, remaining steady between 0.15 nm and 0.20 nm.
From the 10 ns simulation, the stability of the binding complex for C2–F14 and TIPE2 is adequate. The protein has fluctuations of less than 0.1 nm (1 Å) for the entirety of the simulation. And the ligand, after 1 ns, fluctuates around 0.15 nm for the remainder of the simulation. There are no large deviations depicted (Figure 11D). The RMSF plot depicts the fluctuation of the amino acid residues of the complexed protein in comparison to the solvated protein. Overall, the MD studies depict the potential binding stability of TIPE2 and C2–F14.
The C3−TIPE2 binding complex finds stabilization around 1 ns before plateauing. C3 fluxes steadily at 0.3 nm for the duration of the simulation (Figure 11C). No deviations are observed in the protein. RMSD analysis of the MD simulations of C3–F25 shows stability of the complex at 4 ns The ligand experiences cycles of fluctuations. From the start to 4 ns, the ligand fluctuates between 0.35 nm and 0.6 nm. It stabilizes at 4 ns and stays at 0.45 nm, with a slight drop and recovery between 6 ns and 7 ns.
4. Conclusions
TIPE2, a transport protein for PIP2 and PIP3, controls the formation of the leading and trailing edge of a polarized leukocyte leading to the sustainment of chronic inflammation, an environment known to support tumor growth. Thus, we attempted to find a small molecule inhibitor for the protein as a therapeutic option for solid tumor cancers. Other than the traditional structure-based method [29,30], we have conducted a structure-based in silico screening, incorporating fragment-based design techniques, to identify potential TIPE2 inhibitors. We docked 350,000 molecules with TIPE2 using AutoDock Vina and found three potential hit compounds. Due to the large cavity of TIPE2, we attempted to use a pseudo-fragment-based approach, linking fragments to the hit compounds to increase the width of the potential inhibitors and occlude the binding cavity while simultaneously increasing protein–ligand interactions to induce a higher binding affinity. Compound C2–F14 provided us with the highest binding affinity at −12.7 kcalmol−1. Molecular dynamic simulations, conducted with GROMACS, demonstrate a stable binding complex after 1 ns. The computed partition coefficient and TPSA are good, however aqueous solubility could be improved. Overall, we have demonstrated the potential in the development of a small molecular inhibitor for the protein TIPE2.
Author Contributions
All authors contributed to the study’s conception and design. Conceptualization, Y.H.C. and H.-F.J.; methodology, J.W. and K.E.; data curation, J.W. and K.E.; writing—original draft preparation, J.W. and K.E.; writing—review and editing, J.W., K.E., Y.H.C. and H.-F.J.; supervision, Y.H.C. and H.-F.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
All data are available from the manuscript.
Acknowledgments
The work reported here was run on hardware supported by Drexel’s University Research Computing Facility.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Sethi, G.; Shanmugam, M.K.; Ramachandran, L.; Kumar, A.P.; Tergaonkar, V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012, 32, 1–15. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Gattinoni, L.; Powell, D.J., Jr.; Rosenberg, S.A.; Restifo, N.P. Adoptive immunotherapy for cancer: Building on success. Nat. Rev. Immunol. 2006, 6, 383–393. [Google Scholar] [CrossRef]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Ruegg, C. Leukocytes, inflammation, and angiogenesis in cancer: Fatal attractions. J. Leukoc. Biol. 2006, 80, 682–684. [Google Scholar] [CrossRef]
- Lou, Y.; Liu, S. The TIPE (TNFAIP8) family in inflammation, immunity, and cancer. Mol. Immunol. 2011, 49, 4–7. [Google Scholar] [CrossRef]
- Karin, J.A.D.F.M.M. NF-jB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar]
- Sun, H.; Gong, S.; Carmody, R.J.; Hilliard, A.; Li, L.; Sun, J.; Kong, L.; Xu, L.; Hilliard, B.; Hu, S.; et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell 2008, 133, 415–426. [Google Scholar] [CrossRef]
- Oho, M.; Nakano, R.; Nakayama, R.; Sakurai, W.; Miyamoto, A.; Masuhiro, Y.; Hanazawa, S. TIPE2 (Tumor Necrosis Factor α-induced Protein 8-like 2) Is a Novel Negative Regulator of TAK1 Signal. J. Biol. Chem. 2016, 291, 22650–22660. [Google Scholar] [CrossRef]
- Fayngerts, S.A.; Wang, Z.; Zamani, A.; Sun, H.; Boggs, A.E.; Porturas, T.P.; Xie, W.; Lin, M.; Cathopoulis, T.; Goldsmith, J.R.; et al. Direction of leukocyte polarization and migration by the phosphoinositide-transfer protein TIPE2. Nat. Immunol. 2017, 18, 1353–1360. [Google Scholar] [CrossRef]
- Yan, D.; Wang, J.; Sun, H.; Zamani, A.; Zhang, H.; Chen, W.; Tang, A.; Ruan, Q.; Yang, X.; Chen, Y.H.; et al. TIPE2 specifies the functional polarization of myeloid-derived suppressor cells during tumorigenesis. J. Exp. Med. 2020, 217, e20182005. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC.: New York, NY, USA, 2015.
- BIOVIA Dassault Systèmes. Discovery Studio Visualizer, 21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
TIPE2 (A) side, (B) top, and (C) slice.

Figure 2.
Structure of PIP2 (top left) and PIP3 (bottom right).

Figure 3.
Hit Compounds C1–C3.

Figure 4.
C1. (A) Sliced side view of C1 in binding pocket. (B) Sliced top view of C1 in binding pocket. (C) Two-dimensional representation of interactions between C1 and TIPE2 protein residues. (D) Three−dimensional interactions (except van der Waals). (E) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.
Figure 4.
C1. (A) Sliced side view of C1 in binding pocket. (B) Sliced top view of C1 in binding pocket. (C) Two-dimensional representation of interactions between C1 and TIPE2 protein residues. (D) Three−dimensional interactions (except van der Waals). (E) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.

Figure 5.
C2. (A) Sliced side view of C2 in binding pocket. (B) Sliced top view of C2 in binding pocket. (C) Two−dimensional representation of interactions between C2 and TIPE2 protein residues. (D) Three−dimensional interactions (except van der Waals). (E) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.
Figure 5.
C2. (A) Sliced side view of C2 in binding pocket. (B) Sliced top view of C2 in binding pocket. (C) Two−dimensional representation of interactions between C2 and TIPE2 protein residues. (D) Three−dimensional interactions (except van der Waals). (E) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.

Figure 6.
C3. (A) Sliced side view of C3 in binding pocket. (B) Sliced top view of C3 in binding pocket. (C) Two−dimensional representation of interactions between C3 and TIPE2 protein residues. (D) Three−dimensional interactions (except van der Waals) of amino acid residues and C3. (E) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.
Figure 6.
C3. (A) Sliced side view of C3 in binding pocket. (B) Sliced top view of C3 in binding pocket. (C) Two−dimensional representation of interactions between C3 and TIPE2 protein residues. (D) Three−dimensional interactions (except van der Waals) of amino acid residues and C3. (E) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C3 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.

Figure 7.
Structure of fragments F1–F30.

Figure 8.
Highest binding C1 linked ligand, C1−F7. (A) Sliced side view of C1−F7 in binding pocket. (B) Sliced top view of C1−F7 in binding pocket. (C) Two-dimensional representation of interactions between C1−F7 and TIPE2 protein residues. (D) Three-dimensional interactions (except van der Waals) of amino acid residues and C1−F7. (E) Interactions of C1−F7 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C1–F7 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.
Figure 8.
Highest binding C1 linked ligand, C1−F7. (A) Sliced side view of C1−F7 in binding pocket. (B) Sliced top view of C1−F7 in binding pocket. (C) Two-dimensional representation of interactions between C1−F7 and TIPE2 protein residues. (D) Three-dimensional interactions (except van der Waals) of amino acid residues and C1−F7. (E) Interactions of C1−F7 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C1–F7 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.

Figure 9.
Highest binding C2 linked ligand, C2–F14. (A) Sliced side view of C2–F14 in binding pocket. (B) Sliced top view of C2–F14 in binding pocket. (C) Two-dimensional representation of interactions between C2–F14 and TIPE2 protein residues. (D) Three-dimensional Interactions (except van der Waals) of amino acid residues and C2–F14. (E) Interactions of C2–F14 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C2–F14 in binding pocket. Surface area of binding pocket visualized by H-bond affinity.
Figure 9.
Highest binding C2 linked ligand, C2–F14. (A) Sliced side view of C2–F14 in binding pocket. (B) Sliced top view of C2–F14 in binding pocket. (C) Two-dimensional representation of interactions between C2–F14 and TIPE2 protein residues. (D) Three-dimensional Interactions (except van der Waals) of amino acid residues and C2–F14. (E) Interactions of C2–F14 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C2–F14 in binding pocket. Surface area of binding pocket visualized by H-bond affinity.

Figure 10.
Highest binding C3 linked ligand, C3–F25. (A) Sliced side view of C3–F25 in binding pocket. (B) Sliced top view of C3–F25 in binding pocket. (C) Two−dimensional representation of interactions between C3–F25 and TIPE2 protein residues. (D) Three−dimensional interactions (except van der Waals) of amino acid residues and C3–F25. (E) Interactions of C3–F25 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C3–F25 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.
Figure 10.
Highest binding C3 linked ligand, C3–F25. (A) Sliced side view of C3–F25 in binding pocket. (B) Sliced top view of C3–F25 in binding pocket. (C) Two−dimensional representation of interactions between C3–F25 and TIPE2 protein residues. (D) Three−dimensional interactions (except van der Waals) of amino acid residues and C3–F25. (E) Interactions of C3–F25 in binding pocket. Surface area of binding pocket visualized by hydrophobicity. (F) Interactions of C3–F25 in binding pocket. Surface area of binding pocket visualized by H−bond affinity.

Figure 11.
(A) RMSD plot of C1 and TIPE2 in binding complex and RMFD plot of C1–TIPE2 compared to TIPE2. (B) RMSD plot of C2 and TIPE2 in binding complex and RMFD plot of C2–TIPE2 compared to TIPE2. (C) RMSD plot of C3 and TIPE2 in binding complex and RMFD plot of C3–TIPE2 compared to TIPE2. (D) RMSD plot of C2–F14 and TIPE2 in binding complex and RMFD plot of C2–F14–TIPE2 compared to TIPE2.
Figure 11.
(A) RMSD plot of C1 and TIPE2 in binding complex and RMFD plot of C1–TIPE2 compared to TIPE2. (B) RMSD plot of C2 and TIPE2 in binding complex and RMFD plot of C2–TIPE2 compared to TIPE2. (C) RMSD plot of C3 and TIPE2 in binding complex and RMFD plot of C3–TIPE2 compared to TIPE2. (D) RMSD plot of C2–F14 and TIPE2 in binding complex and RMFD plot of C2–F14–TIPE2 compared to TIPE2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Docking scores of C1–C3 with TIPE2 as well as the two natural ligands, PIP2 and PIP3. Also included are the masses of the ligands, calculated solubility, and partition coefficient.
Table 1.
Docking scores of C1–C3 with TIPE2 as well as the two natural ligands, PIP2 and PIP3. Also included are the masses of the ligands, calculated solubility, and partition coefficient.
| Compound | Binding Affinity (kcal mol–1) | Molecular Weight (g mol–1) | Solubility (ESOL) | Partition Coefficient (M logP) |
|---|---|---|---|---|
| C1 | −8.7 | 419.49 | −1.57 | 1.14 |
| C2 | −8.8 | 365.47 | −4.39 | 3.40 |
| C3 | −8.9 | 363.47 | −3.48 | 2.49 |
| PIP2 | −7.0 | 1014.01 | −7.90 | 3.07 |
| PIP3 | −7.0 | 1092.98 | −7.57 | 4.63 |
Table 2.
Binding affinities of hit compounds (C1–C3) linked to their respective top ten binding fragments (F1–F30).
Table 2.
Binding affinities of hit compounds (C1–C3) linked to their respective top ten binding fragments (F1–F30).
| Compound | Binding Affinity (kcal mol–1) | Molecular Weight (g mol–1) | Solubility (ESOL) | Partition Coefficient (M logP) | TPSA (Å2) |
|---|---|---|---|---|---|
| C1–F1 | −8.0 | 821.97 | −6.34 | 1.08 | 159.28 |
| C1–F2 | −10.1 | 823.02 | −7.50 | 1.58 | 166.73 |
| C1–F3 | −10.2 | 831.98 | −7.38 | 1.82 | 157.79 |
| C1–F4 | −9.0 | 827.95 | −6.89 | 0.47 | 182.52 |
| C1–F5 | −10.3 | 804.90 | −5.77 | 0.53 | 187.23 |
| C1–F6 | −9.5 | 782.89 | −5.58 | 1.54 | 174.81 |
| C1–F7 | −10.9 | 793.96 | −7.34 | 1.82 | 163.93 |
| C1–F8 | −9.0 | 815.92 | −7.27 | 0.58 | 153.14 |
| C1–F9 | −10.9 | 799.37 | −7.54 | 2.86 | 135.82 |
| C1–F10 | −9.1 | 822.88 | −6.72 | 1.63 | 153.49 |
| C2–F11 | −10.8 | 619.80 | −6.68 | 2.79 | 96.07 |
| C2–F12 | −11.1 | 658.77 | −6.83 | 3.15 | 108.96 |
| C2–F13 | −10.0 | 654.80 | −6.80 | 2.97 | 108.96 |
| C2–F14 | −12.7 | 651.71 | −6.73 | 2.62 | 135.93 |
| C2–F15 | −10.7 | 642.79 | −6.34 | 2.87 | 98.1 |
| C2–F16 | −10.7 | 653.82 | −7.41 | 3.37 | 93.17 |
| C2–F17 | −11.4 | 615.77 | −6.56 | 2.95 | 96.07 |
| C2–F18 | −10.3 | 657.80 | −7.05 | 3.04 | 106.31 |
| C2–F19 | −11.0 | 609.74 | −6.23 | 3.64 | 92.31 |
| C2–F20 | −10.6 | 660.85 | −5.91 | 3.29 | 95.88 |
| C3–F21 | −10.7 | 660.82 | −8.59 | 5.18 | 70.08 |
| C3–F22 | −9.3 | 657.75 | −7.31 | 3.51 | 108.41 |
| C3–F23 | −10.6 | 671.82 | −7.69 | 4.35 | 107.97 |
| C3–F24 | −9.4 | 669.85 | −7.79 | 4.50 | 93.87 |
| C3–F25 | −11.3 | 669.85 | −7.73 | 4.50 | 93.87 |
| C3–F26 | −9.9 | 674.76 | −7.94 | 4.43 | 117.79 |
| C3–F27 | −10.6 | 534.60 | −5.49 | 2.95 | 88.84 |
| C3–F28 | −9.8 | 572.71 | −5.68 | 4.29 | 70.08 |
| C3–F29 | −8.6 | 554.72 | −5.51 | 3.94 | 70.08 |
| C3–F30 | −10.6 | 669.85 | −7.95 | 4.5 | 93.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wilson, J.; Evangelou, K.; Chen, Y.H.; Ji, H.-F. In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer. Sci 2023, 5, 39. https://doi.org/10.3390/sci5040039
AMA Style
Wilson J, Evangelou K, Chen YH, Ji H-F. In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer. Sci. 2023; 5(4):39. https://doi.org/10.3390/sci5040039
Chicago/Turabian StyleWilson, Jerica, Katerina Evangelou, Youhai H. Chen, and Hai-Feng Ji. 2023. "In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer" Sci 5, no. 4: 39. https://doi.org/10.3390/sci5040039