
Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect



1
Federal Research Center of Problems of Chemical Physics and Medicinal Chemistry RAS, Academician Semenov Avenue 1, 142432 Chernogolovka, Russia
2
Resonance Systems GmbH, 73230 Kirchheim unter Teck, Germany
3
School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK
*
Author to whom correspondence should be addressed.
J. Compos. Sci. 2023, 7(4), 168; https://doi.org/10.3390/jcs7040168
Submission received: 12 March 2023
/
Revised: 31 March 2023
/
Accepted: 10 April 2023
/
Published: 17 April 2023
(This article belongs to the Section Biocomposites)
Abstract
:In 2020, we published a review on the study of semi-crystalline thermoplastic polyurethane elastomers and composites based on the shape memory effect. The shape recovery ability of such polymers is determined by their sensitivity to temperature, moisture, and magnetic or electric fields, which in turn are dependent on the chemical properties and composition of the matrix and the nanofiller. Nanocellulose is a type of nanomaterial with high strength, high specific surface area and high surface energy. Additionally, it is nontoxic, biocompatible, environmentally friendly, and can be extracted from biomass resources. Thanks to these properties, nanocellulose can be used to enhance the mechanical properties of polymer matrices with shape memory effect and as a switching element of shape memory. This review discusses the methods for producing and properties of nanocellulose-based thermo-, moisture-, and pH-sensitive polyurethane composites. The synergistic effect of nanocellulose and carbon nanofillers and possible applications of nanocellulose-based thermoplastic polyurethane biocomposites with shape memory effect are discussed. A brief description of nanocellulose terminology is also given, along with the structure of shape memory thermoplastic polyurethanes. There is significant interest in such materials for three primary reasons: the possibility of creating a new generation of biomaterials, improving the environmental friendliness of existing materials, and exploiting the natural renewability of cellulose sources.
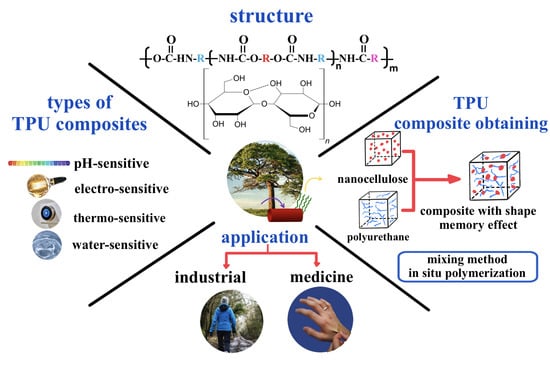
1. Introduction
Segmented thermoplastic polyurethanes (TPUs) represent an important class of adaptive, or smart, shape memory materials (SMMs) [1,2,3,4]. Fundamentally, TPUs are physically crosslinked semi-crystalline block copolymers. They consist of soft polyether or polyester macroglycol (polyol)-based segments and hard segments formed by the reaction of a diisocyanate with low molecular weight diols (and/or diamines) referred to as chain extenders. The thermodynamic incompatibility of soft and hard segments results in a phase-separated structure leading to adaptive properties of the bulk material. Soft segments may exhibit switching behavior (responsible for shape fixing) when the temperature is varied through the glass transition Tg [5,6] or melt Tm [7,8,9] temperatures. The hard segments provide the elasticity of the matrix that is required for shape recovery thanks to the presence of physical cross-links of hydrogen bonds. Improvement and modification of the adaptive properties of TPUs can be achieved either by changing the ratio of soft [10,11,12] and hard segments [1,13,14] or by introducing nanoscale fillers, such as silicon [2] or carbon nanoparticles [15,16,17]. In the last decade, researchers have been particularly attracted to nanoscale particles from natural sources to reinforce polymers or give them new functional properties.
Cellulose is the most abundant renewable and stable biopolymer in nature, surpassing the annual industrial output of all synthetic polymers. Cellulose materials are actively used to create a variety of nanostructures such as nanoparticles of cellulose (nanocellulose) which can be categorized based on their structures as nanocrystals, nanofibrils, and nanocomposites. Nanocellulose is of significant scientific and practical interest due to its optical and mechanical properties, chemical reactivity, biodegradability, recyclability, renewability, and low thermal expansion coefficient. Nanocellulose, however, does have some disadvantages that thus far have limited its widespread application. These include incompatibility with hydrophobic polymers and undesirable moisture absorption. To expand the range of applications of nanocellulose, additional functionality is being explored through covalent modification, blending, cross-linking, polymerization, or a combination of these.
The individual properties of nanocellulose and TPUs can be favorably combined in composite materials to provide significant advantages, especially with regard to mechanical and adaptive properties, biocompatibility, and biodegradability. TPU nanocomposites can contain nanocellulose as a reinforcing nanofiller that enhances the physico-mechanical and relaxation characteristics of the material [18,19]. These enhancements can be attributed to the formation of a percolation network in the polymer matrix and strong hydrogen bonds between the nanocellulose which help to dissipate energy. Thanks to these percolated hydrogen bonding networks, new nanocellulose-based TPU materials are being developed which are able to respond to water [20,21,22] and changing pH levels [23,24]. Nanocellulose can also improve the photothermal, electrical, mechanical, and other functional properties of hybrid TPU materials containing carbon nanoparticles [25,26] or metal nanoparticles [27] by increasing the stability of dispersions in water and organic media as well as the adhesion of these nanofillers to the polymer matrix. In this way, biocomposites based on TPU and nanocellulose, owing to the unique properties of the matrix and renewable sources of nanoparticles, open new horizons in science and biomedicine and industry.
There are a small number of review articles in the literature, which are primarily focused on specific stages of production or use such as the following:
- influence of nanocellulose on the mechanical properties of thermoplastic elastomers.
A smaller number of reviews are also presented on their uses, including the following:
- nanocellulose in drug delivery systems [37].
Diisocyanates have shown great potential as surface modifiers of nanocellulose. In 2019, a mini-review [38] presented commonly used strategies to modify nanocellulose using diisocyanates of different structures, along with their advantages and weaknesses. It is important to note that there are still no review publications describing the methods of obtaining TPU nanocellulose composites with different types of shape activation (i.e., sensitive to temperature and humidity) or structure control allowing the control of their mechanical and adaptive properties. Similarly, no generalized studies on the structure–property relationship of TPU composites containing nanocellulose are currently presented in the scientific literature.
In this review, we discuss the most recent developments in the field of biodegradable TPU composites containing a renewable nanomaterial such as nanocellulose. We will discuss producing nanocomposites, regulation of mechanical and adaptive properties, shape memory activation methods, and the applications of hybrid materials based upon them. It is not possible to cover every published work in this growing field. Instead, we aim to describe the current state of research in as much detail as possible and offer ideas for future opportunities with TPU nanocomposites.
2. From Cellulose to Nanocellulose
2.1. Structural Organization of Cellulose
Cellulose is the most abundant renewable polymer in nature and represents about 50 percent of natural biomass with an estimated annual production of about 50 billion tons [39]. It is worth noting that cellulose is the most environmentally friendly and easily obtainable biopolymer since it can be extracted from many plants.
The chemical structure of cellulose (Figure 1a) is a polysaccharide consisting of a linear chain of several hundreds to many thousands of β-(1 → 4) linked D-glucose units [40] (Figure 1b). Every other monomer is rotated by 180° in relation to the adjacent unit around the polymeric axis. Each cellulose chain has directional chemical asymmetry with respect to the ends of the molecular axis: one end has a chemically reducing functionality due to a hemiacetal unit, whilst the other is nominally non-reducing due to a pendant hydroxyl group (Figure 1a) [41]. Cellulose fibrils have a cross-sectional diameter between 10 nm and 450 nm and lengths up to a few micrometers. Variations in size depend on the biosynthesis features in different material sources [42].
Modern theories of the supramolecular structure of cellulose typically use an amorphous-crystalline representation, with macromolecules of cellulose combined into a crystalline elementary fibril (Figure 1c). Elementary fibrils are generally combined into microfibrils, which are also able to form fibers of higher orders [41]. Microfibrils consist of highly ordered micellar sections, each 50–150 nm long, connected by gaps of 25–50 nm in which the arrangement of elementary fibrils is less ordered. Some authors correlate it with the amorphous phase of cellulose [45]. The ability of glucopyranose rings to form intramolecular and intermolecular hydrogen bonds, however, leads to the aggregation of elementary fibrils even in these spaces. This allows us to attribute the most reactive amorphous part of cellulose exclusively to the surface of the crystallites in the interfibrillar pores [46]. The processing of cellulose fibers often changes both the structure and the sorption properties of the polymer [47].
Micelles and spaces between them do not have clear boundaries and are characterized by a smooth mutual transition into each other. Since the length of the cellulose macromolecule significantly exceeds the length of the micelle, each macromolecule can pass through several sections of microfibrils in which the cellulose chains can interrupt and overlap, resulting in higher fiber strength.
Taking into account the developments in the field of structural organization of cellulose, we also proposed a variant of the model [48] in which a macrofibril is formed from four microfibrils, each of which consists of four elementary fibrils (Figure 1d). Elementary fibrils have a hydrophilic surface and a layered internal structure. Because all hydroxyl groups responsible for inter- and intra-molecular hydrogen bonds lie in the equatorial plane (i.e., the plane in which most of the glucopyranose ring atoms are located), they form alternating layers for the central and angular chains [49]. Similar variants of the structural organization of cellulose microfibrils have also been proposed in the literature [50]. The layers interact due to van der Waals forces and relatively weak hydrogen bonds of the C-H···O type [49]. Both intra- and inter-molecular hydrogen bonds are characteristic of a single monomer (Figure 1a).
2.2. Morphology and Methods of Isolation of Nanocellulose
Cellulose nanoparticles, i.e., nanocellulose, have recently attracted great interest in science and industry. They represent a new class of renewable nanomaterials (NMs) due to their unique mechanical properties combined with nanoscale crystal and fiber morphology, chemically tunable surface, and biodegradability.
As the demand for high-performance materials increases, so has the need for nanocellulose. The growing number of published articles on nanocellulose, particularly those including cellulose nanocrystals (CNCs), is shown in Figure 2 and confirms the great research interest which is being generated by these NMs [51,52,53,54].
2.2.1. Nanocellulose Nomenclature
When analyzing the publications on cellulose nanomaterials (CNMs), it can be observed that several terminologies are used to describe them, leading to potential misunderstanding and ambiguity. Since there is still a dispute in the nomenclature of nanocellulose, it is important to perform a general formalization of the conceptual apparatus for the nanocellulose family and try to use the correct terms based on their morphology, size, and processing method. Several years ago, the Technical Association of the Pulp and Paper Industry (TAPPI) created a nanotechnology division to unify the nomenclature of cellulosic nanomaterials. A draft standard was developed for TAPPI WI 3021 [55]: Table 1 shows the standard terms and their definition for cellulosic nanomaterials. Broadly speaking, CNMs can be divided into nanostructured materials and cellulose nano-objects, which differ in shape and size (Figure 3). Nanostructured materials include the following:
- Cellulose MicroCrystals (CMCs) (e.g., MicroCrystalline Cellulose or MCC) which are a type of cellulose nanostructured material and are approximately 10–15 μm in diameter, contain mostly crystalline regions, and are composed of aggregated bundles of cellulose chains;
Cellulose nano-objects include nanocellulose which, depending on the morphology, particle size, and method of isolation, is divided into three types [29,39,58]:
- Cellulose NanoCrystals (CNCs), so-called crystalline cellulose fibers [53], which are mentioned in the literature with different names, including the following:
- o
- cellulose nanowhiskers (CNWs),
- o
- cellulose crystallites,
- o
- nanorods,
- o
- nanocrystalline cellulose (NCC),
- o
- rod-like cellulose crystals.
These names characterize the form of CNCs: needle-shaped, elongated rod-like, or spindle-shaped with high rigidity of crystal fragments [59] which are described in the literature as rod-like cellulose crystals (whiskers, nanowhiskers, nanorods) [60]. CNCs modification with different shapes (or aspect ratios), sizes, and morphologies can be obtained depending on the source and isolation methods [61]. In general, for the purposes of TPU reinforcement, CNCs in the form of rods of various sizes with a diameter of 3–10 nm and a length of 100–500 nm are used. They are extracted from cellulose fibrils by acid hydrolysis with a degree of crystallinity of 54–88% (according to the X-Ray diffraction method) and a stiffness of 138–200 GPa [42].
- Cellulose nanofibrils (CNFs), so-called semicrystalline cellulose fibers, are mentioned in the literature as [29]:
- o
- nanofibrillated cellulose (NFC),
- o
- nanosized fibrillated cellulose,
- o
- cellulose fibrillar fibers,
- o
- nanofibers,
- o
- cellulose nanofibers,
- o
- aggregates of fibrils, and sometimes
- o
- CMFs or MFC.
The CNF sizes described in the literature vary depending on the natural source with diameters from 5 nm to 30 nm and lengths from 100 to 600 nm [55]. There is, however, a difference between the terms CNF and CMF which should be noted. These names should refer to the different sizes of cellulose nanofibers, which are strongly affected by machining and defibrillation. The defibrillation process creates fibers of smaller diameter (width) for CNF (5 to 30 nm) and larger diameter for CMF (10 to 100 nm). However, this distinction is not always taken into account and both terms are used interchangeably in the literature, leading to some confusion [29,55,62]. Compared to CNCs, CNFs have high aspect ratios (length to diameter), high surface areas, and a large number of hydroxyl groups that are easily available for surface modification. CNFs are long, flexible, and entangled nanocelluloses, which can be extracted from cellulose fibrils by mechanical methods involving several passes through a high-intensity homogenizer [63].
- Bacterial nanocellulose (BNC), bacterial cellulose nanocrystals (BCNCs) or bacterial cellulose nanofibrils (BCNFs) are another type of nanocellulose that differ from cellulose nanocrystals and cellulose nanofibrils [64]. In the literature, BNC are referred to as either of the following:
- o
- Bacterial Cellulose (BC) or
- o
- Microbial Cellulose (MC).
Depending on the growth media and the fermentation method, ultrapure cellulose with different physio-chemical characteristics, size, and degree of crystallinity can be obtained. The degree of polymerization (DP) of BNC varies from 300 to 10,000 depending on the culture conditions, additives, and bacterial strains [65]. BCNFs are shaped like twisted ribbons with an average diameter of 10–40 nm and micrometer length with a large surface area [58]. The Young’s modulus of BCNFs are reported to range from 15 MPa [66] to 114 GPa [67] depending on the crystallinity and structural composition of the samples. Ultrarefining of BNC particles after spray drying results in BCNFs of different sizes and shapes [68]. By treating BNC with a mixture of strong acids, needle-shaped crystals of BNC with a length of 622–1322 nm and a diameter of 33.7–44.3 nm have been produced with increased thermal stability without reduced crystallinity [64]. The degree of crystallinity of BNC synthesized by Acetobacter (e.g., A. xylinum) according to the X-Ray diffraction method is between 84% and 88% [42,66,69].
In addition to the above types of nanocellulose, there are Amorphous NanoCellulose (ANC), Cellulose NanoYarn (CNY), and Amorphous cellulose NanoParticles (ANPs). ANC is a different type of nanocellulose, usually in the form of spherical or elliptical particles with a diameter of 80 to 120 nm. It can be obtained by acid hydrolysis followed by ultrasonic disintegration from regenerated cellulose, which, in turn, is obtained directly from cellulose solution by physical dissolution, molding, and regeneration process [28]. ANPs have been shown to have a spherical shape (Spherical NanoCellulose (SNC)) and are characterized by a high degree of pantamorphia, low DP, and an increased content of sulfonic groups [70].
ANCs are completely hydrolyzed by cellulolytic enzymes to form glucose. Concentrated pastes of ANPs can be used to prevent phase separation of aqueous dispersions of various substances thanks to a thickening effect. Low acidity and soft nanoparticles can be used in cosmetic formulations for gentle skin peeling. Moreover, due to the increased content of acidic functional groups, ANPs can immobilize various Therapeutically Active Substances (TASs) containing basic functional groups. The ANP-TAS complexes can be used in products aimed at effective skin care and treatment. ANCs with improved properties such as high availability, improved sorption, and a higher number of functional groups are primarily used as thickeners in aqueous systems and as carriers for bioactive substances [71].
CNY is one of the least studied nanocelluloses with a fiber size of 100–1000 nm. It is often obtained by electrospinning solutions containing cellulose or its derivatives forming mats of tangled long filaments. CNY has found application for use as wound dressings [72]. The DP of CNY is probably close to the DP of conventional hydrate cellulose fibers, i.e., 300–500. Wide Angle X-Ray Scattering studies reveal that the various kinds of CNY have low crystallinity. Since the stretching stage of the amorphized fibers is absent, the formed nanoyarn will have relatively poor mechanical characteristics. The resulting CNY mats are highly porous and can therefore be used as blotting and filtering materials [72]. More recently, Cellulose nanoplatelets (CNPs) formed by entangled cellulose nanofibers 3 nm in diameter were obtained by oxidation under mild conditions. The thickness of such CNPs is about 80 nm [73].
The nomenclature of nanocellulose—cellulose nanocrystals CNCs, cellulose nanofibrils CNFs, and bacterial nanocellulose BNC as described—will be used throughout this work to be consistent with the standard recommendation of the technical association of the pulp and paper industry [55].
2.2.2. Methods of Nanocellulose Extraction
CNCs have recently attracted significant attention from researchers due to their ability to increase the mechanical strength and lower the density of composite materials along with the excellent renewability, environmental friendliness, and biodegradability of nanocellulose [74]. Compared to thin flexible CNFs and BNC, rigid CNCs with relatively low aspect ratios give unique advantages when used as a reinforcing filler thanks to their rod-shaped morphology.
Since a large number of reviews [29,39,58] are devoted to a detailed description of nanocellulose release methods, we will only briefly discuss the primary methods encountered in the development of functional TPU nanocomposites here.
The isolation or release of crystalline domains in the form of cellulose nanoparticles from purified cellulose materials can be performed in the following ways:
Both chemical and biological processes can be optimized to separate CNCs with the necessary thermal stability and dispersibility.
The most frequently reported chemical method is acid hydrolysis using sulfuric acid [63]. This provides the highest yield of rod-shaped CNCs, which are desirable for use in TPU reinforcement. During acid hydrolysis, amorphous (crystalline surface) and paracrystalline (spaces between the crystalline micelles) regions are hydrolyzed preferentially to the crystalline parts of cellulose, which have a stronger resistance to acid, and thus remain undamaged [79]. This method results in the creation of sulfate groups on the CNCs surface and prevents agglomeration promoting good dispersion in water [80]. The process typically involves acid hydrolysis under controlled conditions (time, temperature, and concentration), centrifugation, dialysis, sonication, freezing, and, finally, drying the suspension.
Acid hydrolysis using hydrochloric acid can also generate CNCs in the same form, but the resulting suspension tends to flocculate (clumping of small particles as flakes), limiting its dispersion. Other types of acids, such as formic acid [81] and hydrobromic acid [82], can also be used for CNCs extraction. The use of these acids in the extraction process, however, has disadvantages, such as corrosive activity and environmental hazards associated with high-energy consumption in addition to their propensity to cause decomposition of the crystalline fragments of nanocellulose.
Hydrolysis of cellulose using phosphoric acid [76] has recently been shown to lead to CNCs with higher thermal stability than those isolated by hydrolysis with sulfuric acid. However, this approach is hampered by the need to use large amounts of weak phosphoric acid and gives a lower yield.
Most CNCs extraction takes place using an optimized sequential addition of both weak and strong acids to give a higher yield of nanocellulose. It has been shown that rod-shaped CNCs with improved thermal stability (260–297 °C) and high dispersion stability can be produced by the combined mild acid hydrolysis method [61,78,83].
Two types of cellulose sources are used in the literature for the preparation of TPU nanocomposites with CNCs extraction by acid hydrolysis:
- Advantec grade №1 filter paper, which consists of 100% alpha-cotton cellulose (cotton powders with a particle size of 20 mm, Sigmacell Cellulose Type 20, crystallinity 79%, and an average molecular weight of about 40,000 Da) [84].
- CMCs from Avicel in two main types depending on the CMCs particle size: Avicel PH101 (particle size less than 50 µm) and Avicel PH301 (particle size ~180 µm) [85]. This is most commonly used to obtain cellulose nanocrystals in the development of TPU nanocomposites [86]. It is purified, partially depolymerized cellulose, extracted by acid hydrolysis (chemically active extrusion) of alpha-cotton cellulose, followed by drying and milling [87].
One promising method for the extraction of CNCs from an aqueous dispersion of microcrystalline cellulose is the scalable high-energy ball milling (HEBM) process [78]. The ball mill has many advantages, such as dry and wet grinding capability, temperature control, and scalability in batch and continuous formats. Recently, ball milling methods with solvents (DMF, DMAA) as liquid media have been successfully used to isolate cellulose nanoparticles. This process advantageously results in the preservation of the inherent crystallinity of the particles. Wet conditions are preferred for cellulose micronization with HEBM because the liquid can create a buffer space between the individual particles preventing the damage to their crystalline structure. Micronization of HEBM cellulose under mild conditions yields CNCs with an average aspect ratio between 20 and 26. CNCs also retain both their degree of crystallinity from 85 to 95% and an increased decomposition temperature (Tonset = 230–263 °C). Similarly, milling CMCs in the presence of dilute phosphoric acid was found to result in CNCs with an average aspect ratio of 21 to 33, high crystallinity (88–90%), and good thermal stability (Tonset = 250 °C). The resulting CNCs have suitable characteristics for improving the mechanical properties of thermoplastic polymers because the higher surface area increases the interaction between the filler and the polymer matrix.
CNFs are extracted by mechanical processing of cellulosic materials, followed by multi-pass homogenization [63]. In order to ease processing, the plant material is pretreated with various methods such as oxidative treatment by acid hydrolysis of phosphoric and sulfuric acids or ionic liquid treatment (for example, imidazolium salts such as 1-butyl-3-methylimidazolium chloride). Then, 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) [88] oxidation can be used as a pretreatment to promote CNF separation and make the nanocellulose surface hydrophobic. When TEMPO nanocellulose is oxidized in the presence of sodium perchlorate and sodium chlorite [89], anionic carboxylates are formed on its surface due to the oxidation of groups at C6 positions, resulting in better dispersion of nanocellulose in water [90]. As such, the obtained nanofibrillated cellulose is approximately 3–4 nm in diameter and a few microns in length with a carboxylic acid surface. TEMPO-oxidized cellulose nanofibers are always uniform width (3–4 nm) with a high aspect ratio and offer applications as transparent and flexible displays, gas-barrier films for packaging, and as nanofiber fillings for composite materials [58].
CNCs and CNFs are extracted from lignocellulosic biomass, while BNC is produced on an industrial scale by the accumulation of low molecular weight sugars by bacteria, especially Gluconacetobacter xylinus, over a period of several days to weeks. BNC is then extracted by static fermentation or fermentation under water [65]. The most efficient production of bacterial cellulose comes from Acetobacters, e.g., A. xylinum, A. hansenii, A. pasteurianus, and Komagataeibacter europaeus [91]. Static fermentation provides BNC with excellent crystallinity and mechanical strength, although long cultivation times and low productivity limit its industrial use. Underwater fermentation leads to a higher yield than static fermentation. Bacterial strains are incubated in a nutrient-rich aqueous environment and produce BNC at the aqueous–air interface as an exopolysaccharide. The elementary fibrils are released through the pores on the cellulose surface, which are then organized and crystallized into microfibrils with a twisted ribbon shape, followed by the formation of a film. BNC is comparable in purity to plant nanocellulose with similar chemical composition to CNCs and CNFs. The properties of the resulting BNC can be modulated by various means such as substrate manipulation, varying culture conditions and operation parameters, and proper selection of the bacterial strain [42].
In summary, the extraction of CNCs for application to TPU composites is based mainly on chemical extraction (hydrolysis) of CMCs with sulfuric or phosphoric acids, in addition to mechanical milling. CNFs, on the other hand, are extracted by oxidization of CMCs. Depending on the process, hydroxyl, carboxyl, sulfate, and phosphate groups are formed on the surface of the resulting nanocellulose. Such nanocellulose is used for the development of TPU composites both in their original form and with subsequent covalent functionalization [42].
3. Enhancing the Mechanical Properties and Shape Memory Effect of Thermoplastic Polyurethanes with Nanocellulose
3.1. Structure of Thermoplastic Polyurethanes
Shape memory polymers (SMPs) have long attracted the attention of scientists because of their extraordinary ability to take different shapes and maintain them until some external action, such as an increase in temperature, brings them back to their original form [92,93,94]. Such polymers belong to the class of thermo-sensitive materials. Of the temperature-responsive adaptive polymers, some of the most promising are TPUs, which have both thermoplastic and elastomeric properties (melting ability, recyclability, and ease of molding). This has led to such materials finding uses in many industrial fields [95,96,97].
TPUs are comprised of a phase-separated block copolymer consisting of thermodynamically incompatible soft segments based on polyether and polyester and hard segments formed by the reaction of diisocyanate with low molecular weight diols (and/or diamines). Figure 4a,b show typical chemical structures and morphologies of TPUs consisting of alternating soft and hard segments interconnected by strong covalent urethane bonds. Hard segments are combined into domains by hydrogen bonds and create a physical network in the polymer. The soft segments ensure the highly elastic deformability of the material. Due to its phase-separated structure, TPU demonstrates the thermo-sensitive shape memory effect (SME), returning to its original shape when heated [98]. Changes between these states are described as fixing (taking on the new shape) and recovery (returning to the original shape).
It is important to note that changes in the mechanical properties of TPUs can occur when the amorphous phase of the soft segment (for example, polyethylene glycol (PEG) or polypropylene glycol (PPG)) decomposes on heating [99,100,101] or as a result of the melting of the crystallizing soft segment (for example, poly(ε-caprolactone) PCL or poly(1,4-butylene)adipate PBA) [11,102,103,104,105]. Thus, the hard segments are responsible for the strength of the material and the fixation of the temporary shape, while the soft segments are responsible for the elasticity and the ability to change into the temporary shape.
Diisocyanates of different types and structures can be used in the synthesis of TPU (Figure 4c) [11,15,106]. This allows the structure of rigid domains to be changed and, as a consequence, changes the mechanical and adaptive properties of the resulting material. Linear aliphatic 1,6-hexamethylene diisocyanate (HMDI) is the most commonly used in the synthesis of TPU as it provides good biocompatibility and no cytotoxicity [107,108,109]. Due to the linear structure of HMDI, a dense network of hydrogen bonds between urethane groups is formed, leading to a high degree of order of the hard segments [14]. This results in strong phase separation between the soft and hard segments, providing the final product with optimized mechanical properties. Along with aliphatic TPU, aromatic diisocyanates such as 4,4′-diphenylmethane diisocyanate (MDI), 2,4-toluylene diisocyanate (TDI), and cycloaliphatic isophorone diisocyanate (IPDI) may be used for the synthesis of TPUs. The symmetrical structure of the aromatic MDI is such that a strong degree of phase separation is achieved between the soft and hard segments, especially at low temperatures [110]. This leads to a regular morphology and consequently high mechanical and adaptive properties of polyurethane materials. Materials based on MDI retain good cytocompatibility and can be used in the medical industry [111,112]. Hard segments based on cycloaliphatic IPDI with different isocyanate group reactivity [113,114] show a weak hydrogen bonding network, which leads to a high proportion of mixing of soft and hard segments, violating the phase-separated morphology [14]. This results in low strength and relaxation characteristics of the material, though, in combination with biodegradable polyols, it gives good biocompatibility and no cytotoxicity, permitting use in medical applications [108,115,116,117]. Aromatic TDI is a particularly interesting diisocyanate since its isocyanate groups in the ortho- and para-positions differ in their reactivity. The isocyanate group in the ortho-position is 5–10 times less reactive than the para-position due to steric hindrance from the adjacent methyl group. This makes aromatic diisocyanate very promising as a crosslinking agent as these groups can selectively react. Due to the high toxicity of aromatic TDI however, polyurethane materials based on it are not suitable for medical applications [116].
The most common chain extender used in the synthesis of TPU is 1,4-butane diol [118] which imparts excellent tensile strength and rebound elasticity to the material with appropriate combination of diisocyanate and polyol groups.
To improve the mechanical and adaptive properties of TPU, a combination of differently structured diisocyanates [1] and chain extenders containing hydroxyl and amine groups can be used [119,120]. This allows the concentration of urethane and urea groups to be regulated, changing the composition of hard segments and thus controlling the rate of shape recovery of thermo-sensitive materials in a targeted approach. The addition of a second polyol PCL, characterized by a lower rate of formation of the crystal phase compared to PBA, allows control of the content and size of crystal domains in TPU. This affects the mechanical properties of the material, changes the switching temperature, and can vary the rate of shape fixation from a few minutes to months [11,106]. By changing the ratio of functional groups, as well as the type and ratio of the crystallizing soft segments, a whole range of TPUs with SME can be obtained with different degrees of stiffness or hardness and response to temperature over a wide range. Depending on the type and composition of soft and hard segments and the chosen preparation method, the relationship between the structure and properties of adaptive TPUs can be extremely diverse. The shape recovery temperature can vary from −30 to 70 °C. A wide operating temperature allows the adaptive properties to be controlled and allows TPUs to find a wide variety of applications [18].
A disadvantage of TPUs is their low stiffness compared to SME metals, resulting in relatively little recovery force when actuated, making them unsuitable for some applications. To overcome this shortcoming, researchers have tried to increase the yield of TPU by adding reinforcing additives such as carbon nanofillers to the polymer matrix [121].
Another interesting behavior of TPU is creep: the time-dependent deformation of materials subjected to constant stress. Creep has both elastic and plastic components and, therefore, can be irreversible after the load is removed, leading to unacceptable deformations and, ultimately, failure of the material. TPU’s relatively low creep resistance is a common drawback that reduces durability and safety, limiting their possible applications. A way to solve this low creep resistance problem is to introduce nanoscale fillers, as demonstrated by Zhen Yao et al. [122], who demonstrated that even low concentrations of nanoparticles significantly improve the creep resistance of polyurethane. It does not, however, increase monotonically with the nanoparticle content because the creep resistance is strongly dependent on the nanoparticle dispersion in the polymer matrix. Due to the low concentrations of nanofillers incorporated in the production of nanocomposites, particle dispersion and wetting are of fundamental importance. These interrelated factors are influenced by processing conditions and the chemical composition of the base material, which also strongly modify the mechanical properties of the composite.
One of the most important factors affecting the reinforcing effect of the filler is the adhesion of the polymer to the surface of the filler. Interfacial adhesion depends on the nature of the bonds at the polymer-solid phase interface. The polymer–filler interaction leads to the development of localized stresses on the surface of the filler particles and determines the deformation and fracture characteristics of the resulting filled polymer.
Nowadays, nanocellulose is an ideal means of improving relaxation properties, thermal and mechanical characteristics, biodegradation period, and hemocompatibility of the polymer matrix. The low cost, low density, high stiffness, and biodegradability of nanocellulose also make its use attractive for many applications. Moreover, nanocellulose has polar groups that can interact with polar polymers such as TPU, resulting in a composite material with good interfacial adhesion [123].
It is expected that the introduction of small concentrations (less than 5 wt%) of nanocellulose particles into TPU will increase Young’s modulus or reduce creep, creating composite materials with increased stiffness without reducing elasticity.
3.2. Influence of the Mixing Method on the Properties of Nanocellulose Enriched Materials
Under suitable conditions, a percolation network of hydrogen bonds may form between the polymer matrix and nanocellulose. The formation of this network is dependent on homogeneous dispersion of the nanofiller and the percolation threshold, which in turn depends on the ratio of nanoparticle sizes and the strength of the filler–matrix interaction. The phenomenon of mechanical percolation is well described in the literature [124,125,126] and will not be covered in depth here. It has been found that the stiffness of composites improves with increasing aspect ratio of nanocellulose [124], causing a decrease in the critical percolation threshold and reinforcing the continuous network. Under these conditions, the basic polymer matrix plays no role in the mechanical stiffness of the material. This corresponds to the greatest mechanical stiffening effect that can be obtained with the introduction of these nanoparticles. For example, the percolation threshold at which TPU nanocomposites based on nanocellulose (TPU/nanocellulose) can demonstrate an increase in tensile strength without sacrificing tensile strain and material stiffness is observed at 1 vol% CNCs. Above this volume fraction, well-dispersed CNCs can contact each other and form large bar-like percolation structures with hydrogen bonds [127]. However, because of the dependence of the percolation threshold on the aspect ratio, the type of cellulose nanocrystals, and the structure of the TPU matrix, this value can be as high as 5 wt%.
TPU/nanocellulose nanocomposites are obtained by mixing nanocellulose or its functionalized derivatives with the polymer matrix or by in situ polymerization (Figure 5). Mixing methods include mixing in solution (dissolution or solvent casting method), sol-gel solvent exchange process, and melt mixing methods, each of which will be discussed in the following subsections.
3.2.1. Solution Method
One of the most common mixing methods is the dissolution or so-called solvent casting method, in which nanocellulose is first dispersed using an ultrasonic bath in a solvent (most often N,N-dimethylformamide (DMF)) and gradually adding TPU before casting and drying (solvent evaporation).
The addition of small amounts of nanocellulose improves the phase separation structure of the TPU matrix and thereby leads to a phase ordering of the soft and hard segments increasing the degree of crystallinity of the soft polyester segments. This makes it possible to control the shape recovery under temperature and strain conditions resulting from cyclic stretching. The improvement in mechanical characteristics, thermal stability, hydrophilic properties of the matrix, and biocompatibility expands possible practical applications of composites based on TPUs [128,129,130].
In the example of polyurethane IROGRAN PS455–203 (Tg = −36.5 °C, Tm = 41 °C) with a crystallizing soft segment, when 1 wt% CNC (isolated by CMCs Avicel PH−101 acid hydrolysis) was added by mixing in solution, an increase (by more than 80%) in the melting temperature of soft segment crystals was observed, indicating a decrease in interphase boundary distance and an increase in crystallinity. In addition, the composite showed an increase in Young’s modulus under tension of approximately 53% and a decrease in creep of around 36% [18].
Hongjie Bi [131] demonstrated improved interphase compatibility between the TPU matrix and CNCs while maintaining the self-repairability of the nanocomposite. Specifically, a mixture containing 1 wt% CNCs, polyurethane (Elastollan S85A), and high molecular weight PCL (Capa6500, Mn ~50,000 Da) was shown to have excellent mechanical properties as demonstrated by its ultimate tensile strength of 31 MPa and elongation at break of 1600%. Khadivi P et al. [85] illustrated the improvement of the shape memory properties as well as the mechanical and thermal stability of a TPU composite based on PEG (Mn ~600 Da), HMDI, and 1,4-butane diol when an optimal concentration of 0.5 wt% CNC (length 100–200 nm and diameter 10–20 nm) is incorporated. The enhancement of the nanocomposite’s properties is due to the induced crystallization and reinforcing effect of CNCs which form a dense network of hydrogen bonds between hydroxyl groups on the nanocellulose surface and the urethane groups of the hard segments. At concentrations lower than 0.5 wt% and higher than 2 wt%, a decrease in the degree of crystallinity of the material was observed due to disordered soft and hard segments associated with the destruction of hydrogen bonds between the urethane fragments [132].
The literature suggests that TPU composites differ significantly when nanocelluloses of different forms are introduced in the same concentration. For example, the introduction of CNFs into a TPU matrix demonstrates a greater improvement in the mechanical properties of the polymer matrix for CNFs than CNCs, which is associated with their higher aspect ratio and flexibility [133,134]. Another article shows that improvement of the mechanical properties of the polymer matrix is also associated with the variable dispersibility of nanocelluloses. Thus, TPU-based composites (synthesized from IPDI, PCL (molecular weight 4000 Da), and 1,4-butane diol) in the presence of CNFs have higher crystallinity and tensile strength than those prepared with CNCs due to better compatibility between CNFs and the TPU matrix [63]. It should, however, be noted that again the degree of crystallinity decreases at higher nanofiller content (more than 5 wt%) [135]. The authors suggest that CNF-induced crystallization of PCL segments is preferable compared to CNCs due to the better dispersibility of the nanofiller in the matrix. As with the CNCs, a rigid network of hydrogen bonds is formed between the CNF’s hydroxyl groups and the urethane groups of the TPU. As the concentration of nanofillers (CNC and CNF) increases, the Young’s modulus and tensile strength also increase, while the tensile strain decreases. The increase in tensile strength observed for the composites in the presence of CNFs was, however, more dramatic compared to the increase observed for the nanocomposites in the presence of CNFs thanks to the formation of a stronger hydrogen bonding network between the matrix and CNFs. The formation of such a network in the case of CNFs is supported by their flexibility due to the high aspect ratio and the presence of amorphous domains along the nanofibers.
The presence of reactive hydroxyl groups on the surface of nanocellulose increases the variety of possible modifications [136,137]. As a result, the stability of dispersions improves, and the thermal stability of polyurethane nanomaterials is increased. Surface modification and pretreatment methods for nanocellulose have now been developed and can be used to target improvement of specific properties. Furthermore, the nanoscale structure is responsible for the exponential increase in hydrogen bonding induced aggregation of these materials.
3.2.2. Nanocellulose Surface Modification for the Production of Polyurethane Nanocomposites with Shape Memory Effect
There are two general methods of modifying the surface of nanocellulose used in the creation of TPU nanocomposites:
- Use of a CNC suspension with surface-active polysaccharides such as chitosan [24].
As a result of these approaches, the CNCs acquire hydrophobic properties which are often important in the development of materials for medical applications.
Chemically modified nanoparticles are dispersed in organic solvents with low polarity and mixed with the polymer solution or directly added to the polymer melt after drying. Two contradictory effects arise from this procedure, however. On the one hand, it improves the dispersion of nanoparticles in a continuous apolar medium, thus optimizing the mechanical properties (particularly strength and compressional modulus), while increasing the thermal stability of the resulting nanocomposite and its ability to respond to multiple external influences. This significantly expands the fields of application of TPU composites. On the other hand, the covalent modification limits the interaction between nanoparticles through the formation of hydrogen bonds of the nanocellulose with the matrix, which is the basis for the outstanding mechanical properties of nanocomposites in the presence of nanocellulose.
Covalent modification of nanocellulose is carried out at the hydrolysis stage to produce the CNCs, and is the most popular method of modifying the nanocellulose surface. The choice of acid also affects the properties of the CNCs obtained: those isolated using sulfuric or phosphoric acid have a negative surface charge due to the introduction of a number of sulfate or phosphate groups on the CNCs surface supporting the electrostatic stabilization of suspensions. The number of sulfate groups depends on the hydrolysis time and concentration of the sulfuric acid. Compared to phosphoric acid, sulfuric acid leads to a much higher surface charge density of the CNCs obtained. Although the literature indicates that phosphate groups introduced during hydrolysis are localized in C2 or C3, there is no evidence for the location of sulfate groups. Detailed descriptions of nanocellulose modification methods and in situ polymerization reactions of their surfaces are the subject of dedicated reviews [136,137].
Cai and co-workers [139] reported an example of chemical surface modification of MCC (particle size 20 mm) using an ionic liquid (1-allyl-3-methylimidazolium chloride) followed by hydrolysis with acetic acid to give improved tensile strength and shape memory TPU composite properties with improved thermal stability and impact strength. Rhodamine B (RhB), a popular fluorescent chromophore with excellent optical properties, was applied as a second modifier to enhance plasticity and provide thermal and photo-responses. The composite was prepared by mixing a polyurethane matrix (Desmopan DP 2795A based on crystallizable PBA, MDI, and 1,4-butane diol), modified MCC (f-MCC), and 0.25 wt% rhodamine in an N-methylpyrrolidone solution (Figure 6a). The results showed that the addition of highly dispersed 5 wt% f-MCC as a crosslinking agent increased impact strength and tensile strength by 1.93 and 2.65 times, respectively (Figure 6e), compared to the neat TPU matrix (Figure 6b). Covalent bonding and cross-linking between f-MCC and hard segments as well as the crystallization of the soft segments (Figure 6a) contributed to good temporal shape fixing (Rf of 98.8%). The chemical network and flexible physical network of hydrogen bonds between rhodamine and urethane groups of the TPU matrix provided plasticity, regulated the degree of phase separation, activated chain mobility, and, subsequently, improved the initial shape recovery. The films with complex geometry easily return to their original shape within 30 s when heated to 60 °C (Rr of 91.4%) (Figure 6c). The shape reversal mechanism of the TPU composite is illustrated in Figure 6b. The strong association between rhodamine and the hard segments of the TPU reduced the proportion of hydrogen bonded carbonyl groups of the hard segments and reduced the number of hard segments dispersed in the soft segments. This increases the mobility of the soft segments in the TPU matrix, leading to a lower glass transition temperature (Tg). On the other hand, the cross-linked network of rhodamine with the hard segment of the TPU matrix is sensitive to temperature changes and plays a dominant role in the shape memory properties of the TPU composite at high temperatures.
Scanning electron microscope (SEM) images of chipped TPU composites before and after cyclic thermomechanical tests (Figure 6d) showed no obvious changes in morphology indicating structural stability. This confirms a simultaneous increase in strength and impact toughness of TPU nanocomposites and lays a significant foundation for their widespread application in, for example, sensors, self-adapting fabric frameworks, and switchable devices.
The nanocellulose surface may be modified with diisocyanates including aliphatic HMDI [140,141], cycloaliphatic IPDI [142], aromatic MDI [128], and TDI [130,143,144,145,146,147] in order to improve the compatibility of nanocellulose with polyurethane matrix and the thermal stability and mechanical properties of the resulting nanocomposites. There are several means of obtaining polyurethane composites by in situ polymerization [38], the most relevant of which will be discussed here.
In the most common approach, one isocyanate group of the diisocyanate reacts with the hydroxyl groups of nanocellulose while the free isocyanate group further reacts with the functional monomer [128,130]. Occasionally, the free isocyanate group is reacted with low molecular weight alcohols to obtain a hydrophobic nanocellulose [143]. Such nanocellulose is then incorporated into a polyurethane matrix by solvent casting to produce a nanocomposite.
In a second variant, the prepolymer is obtained by reacting diisocyanate with macroglycol, whose isocyanate groups are further reacted with the hydroxyl groups of nanocellulose.
In another variant, the terminal isocyanate groups of the diisocyanate react simultaneously with both macroglycol and nanocellulose.
Tian et al. [141] reported the preparation of modified CNC by ureido-pyrimidinone (CNC–UPy) through graft polymerization of the hydroxyl groups of CNCs with isocyanate group of 2-(6-isocyanatohexylaminocarbonylamino)-6-methyl-4[1H]pyrimidinone (UPy–NCO) in DMF solution. As shown in Figure 7, the modified CNC–UPy exhibits a typical rod-like morphology with an average length and diameter of 115 ± 50 nm and 21 ± 6 nm, respectively. It has been shown that a solvent cast composite containing 5 wt% of CNC–UPy exhibits high stiffness, strength, and impact resistance without sacrificing elongation at break compared to pure polyurethane. This is due to the high density of the hydrogen bond network between the TPU matrix and CNC–UPy. The TPU was synthesized from polytetramethylene glycol (PTMG) (Mn ~2000 Da), HMDI, and 1,4-butane diol. It was found that 5 wt% CNC–UPy incorporated into the matrix does not change the mobility of amorphous PTMG chains but prevents the crystallization of soft segments. When the content of CNC–UPs exceeds 5 wt%, the impact strength significantly decreases due to the formation of dimers between CNC–UPs, i.e., it results in a weaker interaction between CNC–UPs and the TPU matrix.
Noormohammadi et al. [129] demonstrated the effect of covalent modification of nanocellulose with an aromatic diisocyanate of symmetrical structure on the structure and mechanical characteristics of TPU nanocomposites. The nanocellulose was modified by the covalent addition of MDI to the CNC’s hydroxyl groups, followed by the interaction of the free isocyanate group with oleic alcohol. Composites obtained by introducing 5 wt% of the Modified CNC (MCNC) into a polyurethane matrix in the melt show an increase in Young’s modulus around 130% without a decrease in impact strength, tensile strength, or elongation at break. An increase in the crystallization temperature and thermal stability of the composites compared to the pure sample was also observed. It has been suggested that the improved properties of the nanocomposite are mainly due to the effective dispersion of modified CNC in the matrix.
Du and coworkers [130] presented an example of covalent modification of CNCs with aromatic TDI followed by interaction of isocyanate groups with 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (Figure 7a) which is an organophosphorus fire retardant to improve the shape memory properties of TPU. TPU was synthesized from PTMG (molecular weight of 2000 Da), TDI, and 1,4-butane diol (BD). The MCNC was found to show good dispersibility in the TPU matrix due to the covalent grafting of urethane groups to the CNC surface by diisocyanate and phosphorus containing additive. The most balanced improvement of crystallization properties (an increase in crystallinity degree and crystallization temperature) due to nucleation of nanoparticles, SME, mechanical, thermal stability, and fire resistance of composites (reduction of heat and smoke emission during combustion) was observed when 3 wt% MCNC was introduced. The composite exhibited rapid material shape recovery within 10 min at 37 °C while its shape fixity and shape recovery ratios (Rf of 94.5% and Rr of 96.8%) were maintained above 90% after 5 cycles (Figure 8c–e). The tensile strength and elongation at break of the composite are 13.1 MPa and 189.3%, respectively. This exciting result demonstrates application of the synthesized composites to create flame-retardant materials.
As mentioned earlier, the ortho-isocyanate group of TDI is 5–10 times less reactive than the para-isocyanate group due to the steric hindrance of the methyl group [148,149]. In such a process, TDI is believed to react with the hydroxyl groups of the CNC surface at the expense of the more reactive para-NCO followed by the interaction of the less reactive ortho-NCO with the desired monofunctional compound or polyol. It is important to note that this reaction is difficult to control because ortho-NCOs are not completely inactive during the first step [150]. This means that some of the TDI on the CNC surface reacts with both ortho-NCO and para-NCO and, therefore, becomes inoperative for subsequent grafting. This may explain the failure of some researchers to graft certain functional monomers onto a surface with CNC in similar quantities to the TDI present, since not all of the TDI molecules have free isocyanate groups [151]. Despite this difference in reactivity, the reaction parameters are significantly influenced by the para-/ortho-selectivity of the isocyanate groups. The disadvantage of these works [128,130] on the modification of the surface of nanocellulose with diisocyanates is the lack of information confirming the selective degree of substitution of para-/ortho-isocyanate groups, which negatively affects the subsequent stoichiometric interaction of the terminal isocyanate groups of the diisocyanate with alcohols or polyols.
It is important to note that despite the great potential of nanocellulose modification with diisocyanates to improve the properties of nanocellulose and composites based on it, there are a number of problems that limit their use; the main problem is that the grafting reaction of diisocyanates to the surface of nanocellulose must take place in a moisture-free environment in order to prevent their hydrolysis and cross-linking to form polyurea. It is also necessary for the nanocellulose to be dispersed into the organic solvents before the reaction starts. This becomes problematic because nanocellulose, especially CNF, tends to aggregate in these solvents, negatively affecting homogeneity; another problem is the difficulty of controlling the reaction between diisocyanates and nanocellulose. Although the intention is for the diisocyanates to react with the surface hydroxyl groups of the nanocellulose through only one of the two isocyanate groups, in reality, this cannot be achieved, resulting in both isocyanate groups becoming non-functional for any subsequent reaction [143,144,145,146]. This problem is made worse by the grafting of diisocyanates of asymmetrical structures such as IPDI and TDI in contrast to diisocyanates of linear structure (HMDI [140,141] and MDI [128]). Furthermore, the molecular flexibility of HMDI compared to TDI increases the ability of both of its isocyanate groups to interact with nanocellulose. Recently, Abushammala [146] developed a simple method to quantify the degree of substitution of ortho-isocyanate groups on the surface of nanocellulose when it reacts with TDI. Using this method, it was possible to optimize the reaction between TDI and nanocellulose to obtain a maximum selectivity of para-/ortho-isocyanate groups of 93%, meaning that 93% of the TDI molecules interact with the ortho-isocyanate groups of the nanocellulose surface. The study also showed that the reaction temperature has a negative effect on the selectivity as it minimizes the difference in reaction kinetics of the para- and ortho-isocyanate groups. The maximum selectivity of 93% can thus be achieved at a temperature of 35 °C with a molar ratio of the isocyanate groups of diisocyanate to the hydroxyl groups of nanocellulose [145]. Another possibility to overcome this problem is to modify the nanocellulose according to the variant involving the initial reaction of the diisocyanate with a functional polyol to form a macrodiisocyanate and then with the nanocellulose via the free isocyanate group. According to this variant of grafting, macrodiisocyanates that have free isocyanate groups will react with nanocellulose, while the other unreacted isocyanate group will be inactive and washed away after the reaction [38].
In the aliphatic IPDI molecule of asymmetric structure, the secondary isocyanate group (-NCO substituent directly linked to the ring) has a higher reactivity than the primary isocyanate group due to steric hindrance of the adjacent methyl group [152]. The reaction selectivity of the secondary isocyanate group can be increased by using dibutyltin dilaurate (DBTDL) as the main catalyst for the urethane formation reaction, while the primary isocyanate groups can be selectively inverted with 1,4-diazabicyclo[2.2.2]octane (DABCO) [113]. This enables the optimization of the selective addition of isocyanate groups to nanocellulose surfaces using two types of catalysts, DBTDL and DABCO [142], the effectiveness of which has been practically demonstrated. These results are in agreement with those of other researchers, who reported 100% conversion of secondary isocyanate groups catalyzed by dibutyl dilaurate tin at 20 °C for a 2:1 stoichiometry of isocyanate groups to hydroxyl groups [153]. Treatment of the CNC surface with IPDI followed by interaction of terminal isocyanate groups with a trifunctional alcohol (ARCOL®LHT-240 based on PPG with molecular weight 700 Da) resulted in an increase in the tensile strength of more than 200% compared to a pure matrix without significant reduction of elongation at break [142].
Imparting hydrophobic properties to the nanocellulose surface can be achieved through modification with cationic surfactants, particularly chitosan, without the formation of covalent bonds [24,154]. Adsorption of the ammonium groups of chitosan on the CNC surface due to electrostatic interactions and hydrogen bonding leads to a reduction of Young’s modulus without changing water absorption of polyurethane composites upon immersion in acidic, alkaline, and neutral media [154].
It is important to note that the solvent casting method has some disadvantages in terms of production speed, production costs and film quality (CNC tends to re-aggregate), and environmental problems due to the high solvent use. Production of polyurethane nanocomposites by mixing in solution, mainly in boiling DMF, has very limited use for large scale technologies due to potential toxicity problems and high melting point (153 °C) as well as the strong hydrogen bonding between the nitrogen atom of the DMF molecule and the hydroxyl group of nanocellulose. It is also challenging to completely remove such solvents by conventional drying processes, even when vacuum assisted, which is essential since residual solvent in the polymer matrix will have a detrimental effect on the properties of the final material. To date, the literature does not contain evidence of complete solvent removal from polyurethane matrices nor does it discuss the impact this has on the ultimate properties of the polymer.
3.2.3. Sol–Gel Solvent Exchange Process
The sol–gel reaction is a process in which a colloidal solution (the so called sol phase) is transformed into a solid gel phase. If the liquid component is then removed through drying, a so called aerogel can be produced. Such an approach can be used to form three-dimensional nanoporous cellulose gels (NCG) [43,155]. This method, based on a sol–gel solvent exchange process, is commonly used to reduce the surface energy of nanocelluloses, thereby improving their dispersibility and compatibility with non-polar media. The main requirements for production using such a process are that the solvent used to dissolve the polymer is miscible with the solvent used to produce an organogel from CNCs and that it does not re-disperse them.
The sol–gel process commences with a homogenous dispersion of CNCs in water or a suitable organic solvent such as N-methylpyrrolidine (NMP) or N,N-dimethylformamide (DMF) creating the sol. Gelation is subsequently induced through solvent exchange with a solvent that is miscible with the first such as acetone, ethanol, methanol, tetrahydrofuran (THF), or isopropanol. The nanocellulose content in this organogel is regulated by the concentration of the initial aqueous nanocellulose dispersion and the ratio to the organic solvent. This gel is then subjected to supercritical drying in alcohol, acetone or CO2, or vacuum freeze drying before being introduced into the polymer matrix [111,156]. More detailed preparation of nanocellulose aerogels and nanocellulose/polymer nanocomposites using sol–gel processes is described in other publications [43,155,157].
So far, only a limited number of examples for such sol–gel processing of CNC-based polyurethane materials have been explored and published. Li K. et al. [158] developed water-sensitive TPU nanocomposites with high cross-linking density and strong interactions between nanocellulose aerogels and polyurethane matrices. Aerogels (NCG) were prepared by replacing the water with acetone and THF, which were then covalently cross-linked with a PEG-based macrodiisocyanate (Mw ~2000 Da) and TDI, followed by the addition of a 3,3-dichloro-4,4-diamino-diphenylmethanelithium chain extender after solvent drying. A supercritical CO2-dried NCG aerogel has been shown to have highly porous networks, consisting of interconnected cellulose nanofibrils with about 20 nm diameter. The introduction of 9–16 vol% NCG aerogel into the hydrophilic TPU matrix leads to an improvement in the mechanical and relaxation properties of the composite.
It is a versatile approach achieving high dispersibility (even in hydrophobic polymers) and transport properties where the CNC hydrogen bonding network plays a vital role. However, solvent swapping is a relatively slow process and, like the solvent casting method, not an environmentally friendly means of producing nanocomposites.
3.2.4. Mixing in the Melt
The third mixing method is the traditional method of mixing in the melt (also known as compounding or reactive extrusion).
In compounding, CNCs can be blended with any thermoplastic polyurethane. The melt compounding process involves high processing temperatures and can therefore only be used to for TPU composites in the presence of CNCs obtained using HEBM rather than acid hydrolysis due to the need for high decomposition temperature and dispersibility. This behavior is usually related to sulfate groups as they can promote dehydration reactions and thereby reduce the thermal stability of the cellulose. The hydrophilicity of CNCs limits dispersion and affects melt workability and interfacial interactions between polymers and fillers. Strong hydrogen bonding between fillers drives a tendency for aggregation and poor dispersion in the polymer matrix during solvent-free processing. To improve the machinability of CNCs in the melt and increase thermal stability, modification of the CNC surface with organic compatibility agents is often used.
Such an approach is discussed by Amin et al. [86], who reported a TPU composite in the presence of HEBM-treated CNC (CNC-MC, length 424 nm, diameter 17 mm, aspect ratio 25) with increased thermal stability. Composites based on CNC treated with sulfuric (CNC-S, length 213 nm, diameter 16 nm, aspect ratio 13) acid show color changes in Texin 990 polyurethane film associated with degradation. This indicates that the production of transparent nanocomposites in large-scale compounding techniques using CNCs treated with sulfuric acid is most likely not feasible. They do, however, report obtaining heat-resistant composites based on TPU of the same Texin 990 grade without changing the optical transparency with CNC-MC and with CNC treated with phosphoric acid (CNC-P, length 270 nm, diameter 26 nm, aspect ratio 10) with no degradation. This has excellent potential for large scale production of transparent nanocomposites. Reinforcement effects in terms of improved tensile strength and impact toughness with no strained effect were observed for nanocomposites with all three CNCs. The improvement in properties was attributed to structural and morphological changes induced by the CNC. At very low concentrations (0.5–1 wt%), the nanocomposites showed improvements in creep modulus and hysteresis properties.
Reactive extrusion uses a standard polymer extruder as a chemical reactor for the synthesis of TPU nanocomposites. The main advantages of the reactive extrusion method are that it is a continuous process, has a large heat transfer area, and is economical. However, instability during polymerization due to large viscosity variations can lead to very low conversion rates or fluctuations in output. Nevertheless, it is considered one of the best methods for producing nanocomposites on a large scale because of its low environmental impact and short processing time.
Garces and coworkers [19] reported the effect of CNC (11.9 nm length, 3.1 nm diameter) concentration on the mechanical properties of thermoplastic polyurethane, DiAPLEX MM4520 (MDI, adipic acid, ethylene glycol, propylene glycol, 1,4-butane diol, bisphenol A, Tg = 45 °C). It was found that the introduction of 0.5–1 wt% CNCs by melt extrusion at 195 °C into the TPU resulted in increased yield strength, impact toughness, bending modulus, and recovery rate. The material has been shown to achieve the best dispersion with the addition of 0.5 wt% CNCs.
Amin et al. [159] demonstrated the scalable introduction of CNCs (diameter 3.45 ± 0. 75 nm and length 497 ± 106 nm, aspect ratio 144) without the use of organic solvents into a TPU matrix synthesized from PTMG (Mw ~41,000 and 98,000 Da), MDI, and 1,4-butane diol, resulting in a significant increase in tensile strength without compromising elastic properties including elongation at break, creep, and hysteresis. It was found that the introduction of nanocellulose, which is a heterogeneous nucleating agent, increases the interface area between the soft segments and the microphase of the hard segments forming nucleation points at the interface. Figure 9 show a TPU (Figure 9a) and a nanocomposite system of TPU/nanocellulose (Figure 9b–d) in which a new smaller microphase is formed from the hard segments. Figure 9a represents the TPU control which may consist of high hydrogen bonding arising from hard segments-hard segments interactions. Increasing the NCO/OH ratio from 1.00 to 1.03 leads to an increase in the nucleation (nucleation point) required to form a new crystalline microphase of the hard segments (Figure 9b–d). The introduction of 0.5 wt% of CNCs at a stoichiometric ratio of 1:1 for NCO:OH functional groups improved the tensile strength of the TPU by up to 43% without reducing the stiffness and elasticity of the TPU matrix. When more than 0.5 wt% CNC is added, phase mixing of hard and soft segments in the TPU matrix is observed, and an increase in the relaxation temperature associated with the formation of hydrogen bonds in the interface region was noted.
Shirole A. et al. [138] demonstrated the introduction of CNC (290 ± 110 nm length and 25 ± 5 nm diameter) containing phosphorylated groups into a TPU matrix by melt mixing. The use of phosphorylated CNCs was motivated by the fact that these particles offer a combination of good dispersibility and high thermal stability. Using the example of Desmopan DP2795A-based on a crystallizable soft switching segment of PBA and hard segments containing MDI and 1,4-butane diol, it was shown that thermomechanical properties can be modified, influencing the crystallization of soft or switching segments or a combination of both by adding a 1 wt% dodecane acid germinator. The introduction of 5–15 wt% phosphorylated CNCs by simply mixing in the melt increased the Young’s modulus from 150 MPa (pure polymer) to 572 MPa (15% CNCs), while mold stability at a given fixation temperature was increased from 47% to 75%. The temperature at which good fixation (>97%) could be quickly achieved was increased from 10 to 25 °C after the addition of 1 wt% dodecanoic acid, which is a nucleating agent for the crystallization of PBA.
In some cases, MCC or BNC are introduced into the TPU chain which, similar to CNCs, increases the strength characteristics of the matrix. For example, the introduction of 0.8 wt% MCC [86] into a TPU chain by extrusion at the macrodiisocyanate stage followed by the addition of a chain extender showed a 28% increase in tensile strength over the original thermoplastic polyurethane and a slight increase in Tg of the soft segment. The slight widening of the endothermic peak in the region of 150–200 °C associated with the melting of paracrystalline hard segments indicates the preferential association of MCC with the hard segment, thereby disrupting the ordering of the hard domains. Introduction of 0.5 wt% BNC by covalent cross-linking with an MDI and soybean polyol-based macrodiisocyanate demonstrates increases in flexural strength and Young’s modulus by 100% and 50%, respectively, compared to the TPU matrix alone [160].
It can be concluded that the blending of TPU with CNCs by means of large-scale production technologies (compounding, reactive extrusion, and molding) without affecting their physical properties, appearance, and elastic material properties is possible when using CNCs obtained by mechanical milling as well as covalent modification of the CNC surface, which gives increased thermal stability.
Composite materials can be prepared by compression molding (temperature 150–200 °C, pressure 100–200 bar, and pressing time 1–4 min) [161] by depositing MFC and CNF between TPU films based on MDI, 1,4-butane diol, and polyether with Tg = −48 °C and Mw ~30,000 Da. The results showed that both types of nanocellulose reinforce the TPU matrix and improve heat resistance. The introduction of 16.5 wt% CNFs to the polyurethane matrix increased the tensile strength by almost 500% and stiffness by 3000% compared to pure TPU.
3.2.5. In Situ Polymerization Method
The limitations discussed in the previous methods are partially alleviated by in situ polymerization. The in situ polymerization method consists of the pre-dispersion of nanocellulose in monomers (with or without solvent) followed by polymerization or curing to produce nanocomposites. However, for large-scale production, the production of polyurethane nanocomposites in solution is limited by the use of large quantities of solvents, which causes environmental problems and significantly increases the cost of producing nanocomposites to unacceptable levels. Despite these disadvantages, composites obtained by in situ polymerization have improved mechanical properties at low filler concentrations such as tensile strength and Young’s modulus, without reducing the elasticity of the matrix.
The introduction of small concentrations of 0.1–2wt% [127,162] CNCs by in situ polymerization of CNC hydroxyl groups with a macrodiisocyanate based on MDI and PTMG (Mn ~1000 Da) followed by chain extension with 1,4-butane diol affects the phase-separated morphology that depends on forming a network of hydrogen bonds between the soft and hard segments [111,127,162,163]. This leads to improved shape memory properties and a reduction in the Tg of the soft segment. The decrease in Tg for nanocomposites is due to the chemical cross-linking of CNCs to the hard segments of the TPU matrix, resulting in a decrease in the fraction of hydrogen bonds between the urethane groups of the hard segment and an increase in the mobility of the soft segment. An 8-fold increase in tensile strength with a marginal increase in modulus of elasticity is observed due to increased compatibility between matrix and nanofiller thanks to the large interfacial surface area and reduced number of agglomerates [127,164]. A higher amount of 2 wt% CNCs, however, leads to restriction of chain mobility due to chemical reaction between hydroxyl groups of CNCs and isocyanate groups of macro diisocyanate. This increases Tg of the soft block and reduces the mechanical properties and crystallinity of the rigid segment due to disruption of the hydrogen bond network [162].
It should be taken into account that in the works published for TPU, nanocomposites containing low concentrations of nanocellulose [127,165,166,167], where MDI or amorphous polyether segments were used, show a decrease in the degree of crystallinity of the hard segment and thermal stability of TPU matrix. When HMDI or crystallizable polyether segments are used for the synthesis of macrodiisocyanate, an increase in crystallinity of the solid phase is observed where low concentrations (1 wt%) of CNCs are introduced, which limits the orientation and crystallization of the amorphous segments during deformation, giving lower values of strength and elongation at rupture [163].
A non-cytotoxic, biocompatible, and biodegradable hydrophilic composite material was developed [168,169] by in situ polymerization, which can be used to create bone scaffolds (a biomaterial structure that serves as a substrate for tissue regeneration) in tissue engineering. The composite is obtained by covalently crosslinking a TPU (based on PCL with Mn ~2000 Da, HMDI, and 1,4-butane diol), 0.1–1 wt% CNCs (length and diameter are >200 nm and <10 nm), and crosslinking agent (acrylic-urethane diene monomer) based on HMDI and 2-hydroxyethyl methacrylate. Increasing the CNC concentration to 1 wt% improved Young’s modulus (19.6 MPa), tensile strength (447%), hydrophilicity, and biodegradation and reduced elongation at break.
Comparing the increase in Young’s modulus obtained for nanocomposites synthesized by in situ polymerization (CNCs chemically bonded to the polyurethane chain) with those obtained by solvent casting (CNCs physically bonded to the polyurethane chain) [170], it was observed that nanocomposites synthesized by in situ polymerization show a higher improvement in strength properties as Young’s modulus values increase by 35% in comparison to 22%, respectively (with the same CNC content).
4. Hybrid Thermoplastic Polyurethane Nanocomposites Containing Nanocellulose
Recently, new hybrid TPU materials have appeared containing nanocellulose and nanofillers (carbon nanoparticles [34,171] or other metal nanoparticles [172]), which impart various functional properties to the polymer matrix.
The main problem facing TPU nanocomposites containing nanofillers is the formation of agglomerates that affect their adaptive and mechanical properties. As described earlier, nanocellulose is a good dispersant and stabilizing agent; thus, it can be expected that its addition to the TPU matrix along with nanofillers will improve the stability of dispersions in aqueous and organic environments. This, in turn, should increase the functional properties of TPU nanocomposite materials.
Carbon nanotubes (CNTs) are the most common carbon nanofillers applied in TPU matrices since they have unexpectedly high heat and electrical conductivities and high strength, in comparison to bulk carbon. They are tubular structures formed from a single cylindrical sheet of graphene (single-walled carbon nanotubes, SWCNTs, or SWNTs) or several sheets of graphene arranged concentrically (multi-walled carbon nanotubes, MWCNT, or MWNT, which also includes double-walled carbon nanotubes DWCNT [173] and few-walled carbon nanotubes FWCNT [174]). The introduction of low concentrations of 0.5–10 wt% CNTs increases the electrical and mechanical properties of the TPU matrix. However, the key problems associated with the introduction of CNTs are discoloration or deterioration in the appearance of polymer composites [161], as well as poor processability at low concentrations due to the formation of agglomerates, which further reduce optical and electrical properties.
There are many active studies of hybrid materials that are a mixture of nanocellulose and carbon-containing nanofillers—mostly CNTs because they are biodegradable and recyclable. These materials can be synthesized with different properties that can be controlled by changing the combination of nanocellulose and CNTs depending on the desired application.
In the production of hybrid materials, the TPU matrix is mixed with a dispersion of nanocellulose and CNTs obtained by physical mixing in solution or by their covalent modification due to functional groups on the surface of nanocellulose and CNTs. Various forms of composites may then be produced from these, including aerogels, papers, foils, or fibers, depending on the ratio and processing [175,176,177]. Methods for obtaining such materials, characterization of properties, and possible applications are well described in recent reviews [34,178].
Despite the small number of different studies in the field of creating hybrid TPU materials based on a mixture of CNTs and nanocellulose, they are expected to find growing application in various fields such as biomedicine and industry, as shown in Figure 10.
A hybrid TPU composite can be obtained by mixing dispersions of nanocellulose and CNTs with a polymer matrix by solvent casting. For example, by mixing solutions of 1 wt% CNFs (CNF-C, particle size 6.5 nm, length 1000 nm) with 5–8 wt% carboxylated CNTs (diameter 30 and 60 nm, length 10–30 μm) in DMF [25], a hybrid electrically conductive TPU composite (conductivity 0.142 S/m) which changes its shape in water within 20 s was obtained (Figure 11). The presence of hydrogen bonding between the TPU matrix, CNFs, and CNTs contributes to the formation of a three-dimensional network structure which changes shape when the material is in contact with water. The shape fixity ratio induced by water (Rf) and the shape recovery ratio (Rr) were 49.65% and 76.64%, respectively. The tensile strength reached 31.78 MPa with a corresponding strain at break of 904.1% (Figure 11). The maximum increase in mechanical properties was observed for 6 wt% CNTs. The trend of the maximum mechanical properties of TPU composites with the introduction of nanoparticles is in good agreement with other sources [132,179,180,181]. It has been shown that the hybrid composite material can be used to reproduce large deformations and recover its original shape in water more than 1000 times with only minor reduction in material properties. This type of TPU composite, according to Wu et al. [182], can be used in the development of sensors or actuators that respond to water.
As with other TPU composites, the aspect ratio of nanocellulose and CNTs leads to an increase in the mechanical and electrical conductive properties, as well as an improvement in the sensitivity to stretching of the prepared sensors [26].
To improve the hydrophobic properties of CNTs and improve dispersion in organic solvents, CNFs may be oxidized with TEMPO to introduce carboxyl groups before mixing into the TPU matrix. This provides better wetting of the CNFs and, therefore, better dispersion of the mixture of CNFs and CNTs in the TPU matrix, leading to improved mechanical properties. Carboxyl groups at 1.698 mmol/g on the surface of CNFs give enhanced CNT dispersion, and the resulting composite film has a wide range of working deformations (up to 507%) [88]. A similar effect can be seen for Irogran PS455–203 with the introduction of a mixture of carboxylated CNFs and MWCNT, which has been demonstrated in [26]. For a content of 3 wt% CNFs and 3 wt% CNTs in the electrically conductive composite, the tensile strength and maximum elongation at break are 35.2 MP and 986%, respectively. The superior deformation performance of the composite is associated with better dispersion of nanofillers, increased interaction between CNFs and the TPU matrix due to its amphiphilicity, and an increase in the overall aspect ratio of CNTs and CNFs from 50 to 150.
CNCs can act as a template, dispersant, and stabilizing agent in the manufacturing of composites with metal nanoparticles together with CNTs [172]. It is been shown that the introduction of 5 wt% CNCs prevented the aggregation of gold nanoparticles and led to an increase in the photothermal effect (for a 532 nm laser) and SME. An increase in the modulus of elasticity of 31.79% and tensile strength by 22.73% was observed compared to pure TPU. The addition of CNCs also prevented the aggregation of gold nanoparticles, which enhanced the photothermal effect and shape-memory properties of the films.
In these ways, hybrid composite materials can be produced which consist of a TPU matrix, carbon nanofillers (CNTs or metal nanoparticles), and nanocellulose. The synergism in the polymer matrix of two functional nanofillers expands the possibilities of using polymer composites and makes it possible to obtain hybrid functional materials with high electrical conductivity, adjustable thermal and electrical conductivity, adjustable optical transparency, photothermal activity, high adsorption capacity, and shape response in the presence of water. Such fillers can also rearrange within the polymer, offering specific applications such as smart temperature control of electronics, smart building insulation, and temperature-adaptive clothing and can be used in any application where variable thermal conductivity can be beneficial.
5. Thermoplastic Polyurethanes with Nanocellulose as Shape Memory Switching Element
In the previous sections we have considered materials for which the mechanical and shape memory effect have been improved using nanocellulose. In this section we consider specifically the case where the shape memory switching effect is created thanks to the presence of nanocellulose.
5.1. Water-Sensitive Thermoplastic Polyurethane Nanocomposites Containing Nanocellulose
In recent years, water-response polymer nanocomposites have become the most attractive for potential biomedical applications (self-tightening sutures, self-healing stents, and active thermoregulation systems) and for use in wearable applications (smart textiles). Nanocellulose is one of the most promising types of materials that can be obtained by modifying SMP with hydrophilic nanofillers.
Water-sensitive SMPs obtained by incorporating CNCs (modulus 143 GPa, length 26 nm, diameter 2.2 µm) into a rubber copolymer of ethylene oxide and epichlorohydrin through a wetting and drying process are described by Capadona and Weder et al. [183]. The swelling of polymer nanocomposites based on nanocellulose in water and other solvents creating competing hydrogen bonds (DMF, isopropanol) results in the formation of a three-dimensional network of hydrogen bonds between nanocellulose and the solvent and gives a corresponding decrease in the elastic modulus of nanocomposites (creating the «off» state). When the solvent evaporates, strong hydrogen bonds are formed between the CNCs (forming a rigid percolation network), which leads to an increase in the elastic modulus of the nanocomposites (creating the «on» state). Thus, the mechanical properties of nanocellulose-based polymer composites can be selectively controlled by changing the hydrogen bond network between nanocellulose and solvent through a wetting–drying process.
In the previous section, we introduced nanocellulose composites with water-sensitive properties [20,21,111,184]. Such composites are obtained with a simple method of mixing a TPU matrix with nanocellulose in a solution before electrospinning. The mechanism for changing and restoring the shape of a TPU composite with polyethers as a soft segment that reacts to water due to the nanocellulose percolation network is shown in Figure 12 [20,185,186]. Here, the characteristic microstructure of a composite is shown, in which water sensitivity is based on the sorption–desorption of nanocellulose during a wetting–drying («off»–«on» state) process.
When the nanocellulose is introduced into the TPU matrix, a temporary network of hydrogen bonds between the nanocellulose (CNC or CNF) and the TPU is formed in the initial dry film. When the composite swells in water, water molecules can diffuse into the film, and competitive hydrogen bonds are formed between water molecules and hydroxyl groups of the nanocellulose, leading to the destruction of the temporary hydrogen bond network. In this state, the film becomes soft («off») and can be easily shaped by an external force. The material is then stretched to the desired deformed state and fixed thanks to the rigid percolation network of nanocellulose created by the removal of water by drying («on»). This new shape will be retained until the material is rewet and the network of hydrogen bonds between the nanocellulose and the TPU matrix is destroyed, restoring the original shape due to the material’s elasticity. The stiffness difference between hard and soft states can be tuned by changing the polymer matrix, the nanocellulose content, or both to suit the requirements of specific applications. Thus, the described SME mechanism is quite different from the traditional one in which water or other solvents are used as plasticizers to lower the transition temperature (melting or glass transition) of SMPs in order to enable shape recovery to start at or below room temperature [111]. The easy-to-apply programmed sorption and desorption process enables rapid recovery of the shape of TPU materials containing nanocellulose.
Wang et al. [187] demonstrated a water-sensitive TPU composite with high sensitivity and response rate based on this method. The nanocomposite was obtained by mixing polyurethane Texin985 based on PTMG, MDI, and 1,4-butane diol (Mn ~74,400 Da) in DMF with 10–40 wt% CNFs. The performance following the introduction of 30% CNF into the TPU matrix is shown by 10 stretch–recovery cycles in Figure 13 with a high shape recovery ratio (about 95%) in less than 1 min.
Zhu et al. [186] demonstrated the effect of CNC concentration on the manifestation of SME for a composite obtained by mixing a CNC suspension with commercial polyurethane HT751 (Mw ~330,000–600,000 Da, polymethyl methacrylate). CNCs are obtained from CMCs (CAS 51395-75-6) by acid hydrolysis with sulfuric acid followed by freeze drying. Composites containing 1–30 wt% CNCs exhibit shape recovery ratio over 90% after wetting for 10 min by immersion in water and fast form shape fixity ratio over 90% in a pre-deformed state at room temperature by drying at 75 °C.
The combination of a nanocellulose percolation network with the entropy elasticity of the TPU matrix is the basis for fixing the temporary shape during deformation in the dry state and restoring it to the original shape in the wet state. These materials represent one of the most promising areas of science due to the opportunities to use them in tissue engineering and controlled release devices.
A new strategy is currently being developed to combine two different types of switches into one material, allowing the TPU composite to respond to two external stimuli at once—for example, combining thermo- and water-sensitivities where the function of the first switch is performed by a crystallizing soft segment of TPU due to its melting when heated, and the function of the second switch is performed by nanocellulose due to the percolation network in water.
Luo et al. [188] well described the mechanism of SME change for TPU composite based on a crystallizable soft segment PCL (Mn ~4000 Da) and a hard segment based on MDI and 1,4-butane diol with a physical network of hydrogen bonds in the presence of CNCs. The structure of an example of such a nanocomposite, sensitive to moisture and temperature, is illustrated in Figure 14. Successful actuation due to change in temperature and immersion in water was demonstrated with the composites restored from a temporary form to a partial form and then finally back to the original form.
Li et al. [158] developed a similar system by introducing CNFs in the form of hydrogels (NCGs) into the TPU matrix obtained by the solvent replacement method, which is a promising method for obtaining TPU composites with improved mechanical properties. This composite was obtained by in situ polymerizations of a PEG-based macrodiisocyanate (Mw = 2000 Da) with TDI (1:1) followed by the addition of 9–45 vol% CNFs (NCG porous cellulose hydrogels) and a 3,3-dichloro-4,4-diamino-diphenylmethanelithium (MOCA) chain extender (Figure 15). NCGs are three-dimensional structured nanomaterials that hold large amounts of water while retaining their interconnected CNF networks after compression and subsequent solvent exchange with acetone and THF to form CNFs. The porosity of compressed NCG hydrogels ranges from 91% to 55%. Introduction of 9–16 vol% NCG results in improved phase separation between soft and hard segments despite high crosslink density and strong hydrogen bonding. The steric hindrances of the rigid NCG structure, however, significantly restrict the mobility of the soft and hard segments, leading to an increase in the glass transition temperature of the soft segments from −52.6 to −51.8 °C and that of the hard segments from 28.7 °C to 32.5 °C. The introduction of 6 to 45 wt% NCG had a significant effect on the mechanical properties of the nanocomposites. Their tensile strength and Young’s modulus increased to 105 and 894 MPa, respectively, with the introduction of 45 wt% NCG compared to 5.5 and 21 MPa for TPU in the absence of nanocellulose. NCG has been shown to improve the mechanical properties of TPU to a greater extent than CNCs. Thus, the tensile strength and Young’s modulus of TPU/CNC nanocomposites are 14.9 and 334.4 MPa, respectively [189], while TPU/MFC nanocomposites are 26.3 and 27.7 MPa, respectively [139], and TPU/BNC nanocomposites 5.2 and 151 MPa, respectively [160]. The resulting composite has strong water- and thermo-sensitive properties and changes shape under the influence of temperature and during the wetting–drying process.
The composite was shown to retain a temporary shape at 0 °C and quickly restore its original shape within 60 s at 60 °C due to the rigid network structure of CNFs and the elastomeric structure of the TPU matrix. After wetting the composite with water at 37 °C, there was a restoration of the shape of the material within 1 min. After evaporation of the absorbed water, the nanocomposites can be easily deformed into a temporary shape under the action of an external mechanical force resulting from the destruction or transformation of the strong hydrogen bond between the CNFs.
Electrospinning is a technique which has been used for over a century to produce narrow fibers, which are typically deposited into a random mat. Cellulose acetate was first used to produce cellulose fibers as early as 1938 to produce gas mask filters [190]. It is only in recent years, however, that the technique has been employed to produce SME composites with cellulose nanofibers in TPU. In the electrospinning technique, fibers of a few tens to hundreds of nanometers are produced by using a high voltage to force materials through small apertures, effectively extruding materials into fibers. In the work of Tan et al., for example [185], cellulose acetate (Mn ~30,000, 39.8 wt% acetyl) is dissolved for 2 h at room temperature in a binary solvent mixture of acetone/DMAc 3:1 by weight before producing a nanofibrous mat using a commercial electrospinner at 1.0 kV/cm. The mats are then deacetylised by soaking for 30 min in 0.5 mol/L sodium hydroxide/absolute ethanol solution. Solution casting is then used with the nanofibers and HT-751 TPU chips. The resulting composites demonstrated water-driven SME-effects with fixation ratios of up to 70% and recovery ratios over 90% after an initial cycle and provide repeatable behavior over 5 test cycles.
The main works in this area, including composite composition, main properties of their shape memory, and the type of nanoparticles used are presented in Table 2.
5.2. pH-Sensitive Thermoplastic Polyurethane Composites Containing Nanocellulose
Materials that change shape in physiological solutions at a given pH have excellent potential for use in medical applications since a rapid change in the pH value of various organs and tissues is often observed under pathological conditions [191].
To date, few papers have been published describing the manifestation of SMEs in TPU/nanocellulose composites through dissociation and association of hydrogen bonds under the influence of changes in the ambient pH.
To improve the water absorption properties of TPU composites in various pH environments, the CNC surface can be manipulated by covalent modification or modification using cationic surfactants without forming covalent bonds. For example, by modifying the CNC surface with chitosan, Wu and coworkers [24] were able to modify the mechanical properties of TPU composites depending on changes in the pH of the medium, which enables their application in both acidic and neutral environments. Chitosan is a polysaccharide obtained from chitin input by N-deacetylation [192], which dissolves (or swells upon crosslinking) when exposed to an acidic environment. Chitosan is positively charged under acidic conditions (cation sites: 5000 mmol·kg−1) [154] and can be adsorbed on the CNC surface by the driving forces of electrostatic interactions and hydrogen bonds. Decreases in moduli and equilibrium rate of water absorption rates were found for solutions with pH = 4 (1% acetic acid), pH = 13 (2% NaOH), and pH = 7 (distilled water). The ability of the composite to generate force (up to 0.11 N) has potential for applications in water-sensitive switches, sensors, and ultra-low power generation with the use of natural polysaccharides as functional components.
During the covalent modification of CNCs, various functional groups with a positive and negative charge are introduced onto their surfaces. CNCs are typically rich in sulfate ester groups on their surface, which are introduced by sulfuric acid hydrolysis [193]. The total number of anionic centers was reported to be between 47 and 110 mmol/kg with surface charge densities between 0.68 and 0.155 enm2 [193]. The dissociation of sulfate ester groups and hydroxyl groups on the CNC surface is also pH-sensitive, which ensures the pH-sensitive wettability of CNCs.
The CNC surface can also be functionalized with pyridine moieties via hydroxyl substitution of CNCs with pyridine-4-carbonyl chloride and carboxyl groups (CNC-CO2H) via TEMPO-mediated surface oxidation. Examples of the interesting properties of pH-sensitive TPU composites are described in [23,194]. A pH-sensitive composite was obtained by mixing a TPU solution based on a PEG-PCL copolymer, MDI, and 2,2-bis(hydroxymethyl) propionic acid chain extender with 5–20 wt% CNC functionalized with positively charged pyridine fragments (CNC-C6H4NO2). The resulting composite is insensitive to changes in heat and moisture, only displaying SME behavior when the pH changes. At high pH, pyridine is deprotonated, facilitating hydrogen bond interactions between the pyridine groups (and hydroxyl moieties) on the CNCs and the carbonyl groups (and amide groups) of the TPU matrix, while at low pH, protonation of the pyridine moieties reduces the interactions. In comparison, carboxyl-functionalized CNCs have an opposite response to pH changes [195,196], as the surface of carboxylated CNCs is negatively charged. The percolated network of the pH-sensitive TPU composite in the presence of CNC-C6H4NO2 and the process of changing the SME of the polymer material is shown in Figure 16 [23,194].
TDI—2,4-toluene diisocyanateThe values of Rf and Rr (more than 85%) for pH-sensitive TPU materials containing 20 wt% CNC-C6H4NO2 are sufficiently stable and show good repeatability with an increase in the number of cycles despite the decrease in Rr to 65% after the sixth cycle due to the orientation of nanocellulose along the drawing direction. Along with the pH sensitivity of CNCs, the mechanical properties of the composite are also improved [195].
Such pH-sensitive SME composites are promising in the development of biomaterials for medical applications (e.g., drug delivery systems, tissue engineering applications, or identifying infected tissues), where CNCs serve as a block for switching the properties of TPU materials in a physiological environment [24].
6. Application of Composites Based on Thermoplastic Polyurethane and Nanocellulose
Biocomposites based on TPU and nanocellulose are widely available and have good dispersibility, biocompatibility, and minimal cytotoxicity, which has seen their use in diverse applications. Our description of the biological properties of TPU was presented in the previous review [98] and goes beyond the present work.
In addition to the creation of multifunctional materials that react to heat, water, and changes in the pH of the environment, it is possible to significantly expand the prospects for using such materials in industrial applications through the addition of carbon and metal nanofillers (electrically conductive, photo-sensitive, and thermo-sensitive matrices), leading to opportunities in smart fabrics, fire-resistant materials, strain gauges, and biomedicine amongst others.
Currently, researchers have focused on the production of functional textiles from SME thermo-sensitive TPUs due to the wide transition temperature range, ease of manufacture, and the ability to produce fibers based on them using various production methods such as melt spinning and electrospinning, giving smart textiles which perceive, interact, and adapt to environmental influences.
Examples of thermally sensitive TPU composites based on CNCs with hydrophilic properties and the prospect of using such composites to create smart fabrics with a double response to both environmental temperature and humidity are particularly promising for practical applications [22,196]. Treatment of woolen fabrics with a nanocellulose-based polyurethane composite has been shown to prevent wool curling [22,196]. Korkmaz and co-workers [196] demonstrated that woolen fabrics modified with TPU nanocomposites (containing 5–20% CNCs) have a thermo- and moisture-sensitive response providing dynamic air permeability (the ability of the material to expel body evaporation) and sweat absorption at different temperatures and ambient humidity as well as excellent mechanical properties and the ability to prevent felting. This TPU nanocomposite (Figure 17) was obtained by mixing commercial polyurethane MM-3520®TPU (prepared from MDI, propylene glycol, PEG, 1,4-butane diol, and bisphenol A, Tg = 32.12 °C) with CNC (length 150–200 nm and width 20 nm, crystallinity 98.98%).
The introduction of CNCs modified with urethane-phosphorus-containing flame-retardant units (MCNC) into the TPU matrix holds promise for the use of such polymers as flame-retardant materials [130]. Such a composite material can be obtained using the solution method in DMF by mixing TPU based on PTMG (Mn ~2000 Da), TDI, and 1,4-butane diol with 3 wt% MCNC. This composite showed an improved limiting oxygen index value (23.8%) compared to the neat TPU matrix. In the vertical combustion test, self-extinguishing was observed in less than 30 s after the flame source was removed, while the composite burned with small flame drops. Together with the good dispersion of cellulose nanoparticles in a TPU matrix, the presence of nitrogen- and phosphorus-containing units on MCNCs can catalyze the formation of a phosphorus-reinforced carbonaceous block (solid phase) and promote the production of non-combustible gas products (gas phase) causing a self-extinguishing flame (Figure 18). The carbon barrier created can significantly reduce the transfer of oxygen and heat and the release of volatile products and, therefore, protect the underlying TPU composite.
Hybrid TPU composites based on nanocellulose can be used as strain gauges [25,26] with improved electrically conductive properties due to a better dispersion of CNTs in the polymer matrix. For example, it was shown [26] that with an increase in the CNF content from 0.5 to 1 and 3 wt%, the conductivities went from 0.0012 S/m to 0.0313 S/m and up to 0.28 S/m, respectively. This increase in electrical conductivity is due to the improved dispersion of the CNTs within the TPU matrix. Guanzheng Wu et al. [25] demonstrated the operation of a strain gauge based on a hybrid material of polyurethane BT-70ARYU, CNFs, and carboxylated MWCNT used for finger and arm flexion. The authors showed that such a gauge can effectively respond to human movement whilst the water-sensitive TPU material gives the sensor stability to a high number of cycle repetitions in a humid environment. The combination of responses to water and electricity will allow such materials to be used to develop new generations of water-sensitive sensors, actuators, or biomedical devices.
Similar materials containing electrically conductive nanoparticles have been developed for use as functional paper substrates in flexible electronics and information storage devices. Their properties depend on the size of the cellulose nanofibers and the amount of electrically conductive additive. The TPU composite was prepared by mixing a solution of polyacrylate urethane with 10% α-hydroxyketone to form a polyurethane gel followed by the introduction of a cellulose film with UV curing [197]. The cellulose film was obtained by covalent modification of a CNF-CNT sandwich. The successful production of highly transparent cellulose sheets impregnated with TPU/CNT composite with low permeability and electrically conductive properties (with a content of 5 wt% CNTs, the electrical conductivity was 0.0491 S/m) had a tensile strength of 4.08 MPa, a Young’s modulus around 261.19 Mpa, and elongation at break equal to 3.43%.
Designing scaffolds in tissue engineering from biocompatible materials is one of the most difficult problems in regenerative medicine since it is highly critical to the outcome. These materials must be biocompatible, provide a surface to which cells can successfully adhere, and should promote the ongoing growth of the cells. One of the problems with existing TPU materials used in the medical industry is their hydrophobicity. To overcome this problem, new biocompatible and hydrophilic TPU composites are being developed [168] by fabricating semi-interpenetrating polymer networks using cross-linking obtained by the reaction of HMDI with 2-hydroxyethyl methacrylate in the presence of CNC. The composite (semi-IPN) was prepared by mixing DMF into TPU based on PCL (Mn ~2000 Da), HMDI, and 1,4-butane diol with CNCs (length 100 nm and diameter 7 nm) alongside an acrylic urethane crosslinking agent. Stained microscopy of fibroblast cells (SNL76/7) confirms the results of the biocompatibility of TPU films in the presence of 0.1–1.0 wt% CNCs. Such composites have good biodegradation, degree of hydrophilicity, and mechanical properties and are non-cytotoxic. Studies in this field have also been published in other sources [162,169,198], indicating the prospects for the development of TPU composites based on nanocellulose in regenerative medicine.
7. Conclusions
In this review, we have presented the results of recent advances in the synthesis, modification, and application of adaptive composites based on thermoplastic polyurethanes and a renewable source of nanoparticles: nanocellulose (CNCs and CNFs). An important advantage of the TPU matrix class is the customization of the properties of polymeric materials for specific applications through thermodynamically incompatible soft and hard segments. The use of nanocellulose as nanoscale additives for TPU matrices can significantly improve the mechanical properties of the polymer composite and give it resistance to biodegradation.
It is worth noting that the final mechanical and adaptive properties of TPU composites, such as stiffness, strength, creep, and adhesion depends on many factors including the nanocellulose type, method used for isolation, aspect ratio, and mechanical threshold of percolation as well as the method used to produce the composite (solvent casting method, in situ polymerization, sol–gel solvent exchange process, compounding) and nanoparticle–matrix compatibility amongst others. The addition of nanocellulose or its derivatives obtained by covalent functionalization of its surface not only improves the mechanical properties and thermal stability of the matrix but also permits creation of composites with double memory responsive to water and changes in the pH of the environment. This creates opportunities for use in industry—for example, in the production of smart textiles, flame-retardant materials, and regenerative medicine scaffolds. The addition of nanocellulose to hybrid TPU composites in the presence of carbon nanofillers improves the conductive properties of the matrix, affecting the shape fixity ratio and shape recovery rate of smart materials by improving the dispersibility of carbon nanoparticles within the matrix. Such materials are showing great promise for the development of sensors—for example, in measuring strain.
It is important to note that, currently, most adaptive TPU composites are produced by solvent casting, which has issues around production speed, cost, and environmental impact as well as its tendency to exhibit nanocellulose reaggregation. There is no evidence of complete removal of the solvent from the polymer and no data on its effect on material properties, which is an area requiring further research. The latest compounding methods (mixing in the melt or high-performance extrusion) are used mainly for industrial scale production and are of little use for low-temperature medical composites in the presence of nanocellulose obtained by acid hydrolysis due to their low thermal stability.
One of the promising ways to obtain TPU nanocomposites is the modification of nanocellulose surfaces with diisocyanates. Despite the claimed potential of isocyanate compounds as chemical linkers and surface modifiers, researchers encounter many problems when isocyanate groups of diisocyanates interact with hydroxyl groups of nanocellulose, including the need for a moisture-free environment, nanocellulose aggregation during solvent exchange, reaction controllability, and self-polymerization of diisocyanates.
The large number of publications in this field, however, leaves no doubt that researchers will be able to overcome these shortcomings, resulting in the widespread introduction of composites based on adaptive TPUs and nanocellulose in countless high-tech fields.
Author Contributions
All authors contributed to the study conception and design for review article. Idea for the article, the literature search, and data analysis: M.G.; writing article: M.G.; review and editing: L.G. and R.H.M.; critically revised the work: L.G. and R.H.M.; editing the article, adapted figures, and graphic abstract: M.G. and A.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. This work was performed by the state task 0089-2019-0012, state registration No AAA-A19-119032690060-9.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are listed in the References, which are openly available on the web.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Baek, S.H.; Kim, J.H. Shape Memory Characteristics of Thermadapt Polyurethane Incorporated with Two Structurally Distinctive Aliphatic Isocyanates. Polym. Test. 2021, 103, 107366. [Google Scholar] [CrossRef]
- Mehrbakhsh, E.; Rezaei, M.; Babaie, A.; Mohammadi, A.; Mayan Sofla, R.L. Physical and Thermo-Mechanical Properties of Shape Memory Polyurethane Containing Reversible Chemical Cross-Links. J. Mech. Behav. Biomed. Mater. 2021, 116, 104336–104347. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S. Temperature Responsive Shape Memory Polyurethanes. Polym. Technol. Mater. 2021, 60, 1491–1518. [Google Scholar] [CrossRef]
- Constante, G.; Apsite, I.; Auerbach, P.; Aland, S.; Schönfeld, D.; Pretsch, T.; Milkin, P.; Ionov, L. Smart Mechanically Tunable Surfaces with Shape Memory Behavior and Wetting-Programmable Topography. ACS Appl. Mater. Interfaces 2022, 14, 20208–20219. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Zhang, H.; Qu, J.; Wang, J.; Bai, Y.; Sai, F. Recyclable Shape-Memory Waterborne Polyurethane Films Based on Perylene Bisimide Modified Polycaprolactone Diol. Polymers 2021, 13, 1755. [Google Scholar] [CrossRef]
- Kim, B.K.; Shin, Y.J.; Cho, S.M.; Jeong, H.M. Shape-Memory Behavior of Segmented Polyurethanes with an Amorphous Reversible Phase: The Effect of Block Length and Content. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 2652–2657. [Google Scholar] [CrossRef]
- Karasu, F.; Weder, C. Tuning the Properties of Shape-Memory Polyurethanes via the Nature of the Polyester Switching Segment. Macromol. Mater. Eng. 2021, 306, 2000770–2000781. [Google Scholar] [CrossRef]
- Huang, Z.; Ban, J.; Pan, L.; Cai, S.; Liao, J. New Star-Shape Memory Polyurethanes Capable of Thermally Induced Recovery and Hydrogen Bond-Self-Healing. New J. Chem. 2021, 45, 8427–8431. [Google Scholar] [CrossRef]
- Nissenbaum, A.; Greenfeld, I.; Wagner, H.D. Shape Memory Polyurethane—Amorphous Molecular Mechanism during Fixation and Recovery. Polymer 2020, 190, 122226–122236. [Google Scholar] [CrossRef]
- Czifrák, K.; Lakatos, C.; Árpád Kordován, M.; Nagy, L.; Daróczi, L.; Zsuga, M.; Kéki, S. Block Copolymers of Poly(ω-Pentadecalactone) in Segmented Polyurethanes: Novel Biodegradable Shape Memory Polyurethanes. Polymers 2020, 12, 1928. [Google Scholar] [CrossRef]
- Gorbunova, M.A.; Anokhin, D.V.; Lesnichaya, V.A.; Grishchuk, A.A.; Badamshina, E.R. Optimization of Structure of Soft Block for Design of Adaptive Polyurethanes. Key Eng. Mater. 2020, 869, 273–279. [Google Scholar] [CrossRef]
- Wu, Y.H.; Wang, C.C.; Chen, C.Y. Effect of the Cyclic Structure Content on Aliphatic Polycarbonate-Based Polyurethane. Polym. J. 2021, 53, 695–702. [Google Scholar] [CrossRef]
- Mohanty, J.; Garg, H.; Gupta, P.; Alagirusamy, R.; Tripathi, B.P.; Kumar, B. Mechanically Strong and Resilient Shape Memory Polyurethane with Hexamethylene Diisocyanate as Mixing Segment. J. Intell. Mater. Syst. Struct. 2021, 32, 733–745. [Google Scholar] [CrossRef]
- Gorbunova, M.A.; Komov, E.V.; Grunin, L.Y.; Ivanova, M.S.; Abukaev, A.F.; Imamutdinova, A.M.; Ivanov, D.A.; Anokhin, D.V. The Effect of Separation of Blocks on the Crystallization Kinetics and Phase Composition of Poly(Butylene Adipate) in Multi-Block Thermoplastic Polyurethanes. Phys. Chem. Chem. Phys. 2022, 24, 902–913. [Google Scholar] [CrossRef]
- Gorbunova, M.; Komratova, V.; Grishchuk, A.; Badamshina, E. The Effect of Addition of Low-Layer Graphene Nanoparticles on Structure and Mechanical Properties of Polyurethane-Based Block Copolymers. Polym. Bull. 2019, 76, 5813–5829. [Google Scholar] [CrossRef]
- Kausar, A. Shape Memory Polyurethane/Graphene Nanocomposites: Structures, Properties, and Applications. J. Plast. Film Sheeting 2020, 36, 151–166. [Google Scholar] [CrossRef]
- Babaie, A.; Rezaei, M.; Sofla, R.L.M. Investigation of the Effects of Polycaprolactone Molecular Weight and Graphene Content on Crystallinity, Mechanical Properties and Shape Memory Behavior of Polyurethane/Graphene Nanocomposites. J. Mech. Behav. Biomed. Mater. 2019, 96, 53–68. [Google Scholar] [CrossRef]
- Auad, M.L.; Contos, V.S.; Nutt, S.; Aranguren, M.I.; Marcovich, N.E. Characterization of Nanocellulose- Reinforced Shape Memory Polyurethanes. Polym. Int. 2008, 57, 651–659. [Google Scholar] [CrossRef]
- Garces, I.T.; Aslanzadeh, S.; Boluk, Y.; Ayranci, C. Cellulose Nanocrystals (CNC) Reinforced Shape Memory Polyurethane Ribbons for Future Biomedical Applications and Design. J. Thermoplast. Compos. Mater. 2020, 33, 377–392. [Google Scholar] [CrossRef]
- Mendez, J.; Annamalai, P.K.; Eichhorn, S.J.; Rusli, R.; Rowan, S.J.; Foster, E.J.; Weder, C. Bioinspired Mechanically Adaptive Polymer Nanocomposites with Water-Activated Shape-Memory Effect. Macromolecules 2011, 44, 6827–6835. [Google Scholar] [CrossRef]
- Qi, X.; Jing, M.; Liu, Z.; Dong, P.; Liu, T.; Fu, Q. Microfibrillated Cellulose Reinforced Bio-Based Poly(Propylene Carbonate) with Dual-Responsive Shape Memory Properties. RSC Adv. 2016, 6, 7560–7567. [Google Scholar] [CrossRef]
- Korkmaz Memiş, N.; Kaplan, S. Production of Thermal and Water Responsive Shape Memory Polyurethane Nanocomposite Filaments with Cellulose Nanowhisker Incorporation. Cellulose 2021, 28, 7075–7096. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Liu, D.; Wang, W.; Liu, Y.; Zhou, S. PH-Responsive Shape Memory Poly(Ethylene Glycol)-Poly(ε-Caprolactone)-Based Polyurethane/Cellulose Nanocrystals Nanocomposite. ACS Appl. Mater. Interfaces 2015, 7, 12988–12999. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Su, Y.; Chen, B. Mechanically Adaptive and Shape-Memory Behaviour of Chitosan-Modified Cellulose Whisker/Elastomer Composites in Different PH Environments. ChemPhysChem 2014, 15, 2794–2800. [Google Scholar] [CrossRef]
- Wu, G.; Gu, Y.; Hou, X.; Li, R.; Ke, H.; Xiao, X. Hybrid Nanocomposites of Cellulose/Carbon-Nanotubes/Polyurethane with Rapidly Water Sensitive Shape Memory Effect and Strain Sensing Performance. Polymers 2019, 11, 1586. [Google Scholar] [CrossRef]
- Xu, S.; Yu, W.; Jing, M.; Huang, R.; Zhang, Q.; Fu, Q. Largely Enhanced Stretching Sensitivity of Polyurethane/Carbon Nanotube Nanocomposites via Incorporation of Cellulose Nanofiber. J. Phys. Chem. C 2017, 121, 2108–2117. [Google Scholar] [CrossRef]
- Hu, Z.; Fu, S.; Tang, A. Fabrication of Light-Triggered AuNP/CNC/SMP Nano-Composites. BioResources 2017, 12, 1982–1990. [Google Scholar] [CrossRef]
- Trache, D.; Tarchoun, A.F.; Derradji, M.; Hamidon, T.S.; Masruchin, N.; Brosse, N.; Hussin, M.H. Nanocellulose: From Fundamentals to Advanced Applications. Front. Chem. 2020, 8, 392–425. [Google Scholar] [CrossRef]
- Santos, R.F.; Ribeiro, J.C.L.; Franco de Carvalho, J.M.; Magalhães, W.L.E.; Pedroti, L.G.; Nalon, G.H.; Lima, G.E.S. de Nanofibrillated Cellulose and Its Applications in Cement-Based Composites: A Review. Constr. Build. Mater. 2021, 288, 123122–123139. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated Cellulose—Its Barrier Properties and Applications in Cellulosic Materials: A Review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Dufresne, A. Nanocellulose: A New Ageless Bionanomaterial. Mater. Today 2013, 16, 220–227. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Xie, Y.; Zhang, K. Functional Nanomaterials through Esterification of Cellulose: A Review of Chemistry and Application. Cellulose 2018, 25, 3703–3731. [Google Scholar] [CrossRef]
- Islam, M.S.; Chen, L.; Sisler, J.; Tam, K.C. Cellulose Nanocrystal (CNC)–Inorganic Hybrid Systems: Synthesis, Properties and Applications. J. Mater. Chem. B 2018, 6, 864–883. [Google Scholar] [CrossRef]
- Miyashiro, D.; Hamano, R.; Umemura, K. A Review of Applications Using Mixed Materials of Cellulose, Nanocellulose and Carbon Nanotubes. Nanomaterials 2020, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Bacakova, L.; Pajorova, J.; Tomkova, M.; Matejka, R.; Broz, A.; Stepanovska, J.; Prazak, S.; Skogberg, A.; Siljander, S.; Kallio, P. Applications of Nanocellulose/Nanocarbon Composites: Focus on Biotechnology and Medicine. Nanomaterials 2020, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Nasseri, R.; Deutschman, C.P.; Han, L.; Pope, M.A.; Tam, K.C. Cellulose Nanocrystals in Smart and Stimuli-Responsive Materials: A Review. Mater. Today Adv. 2020, 5, 100055. [Google Scholar] [CrossRef]
- Raghav, N.; Sharma, M.R.; Kennedy, J.F. Nanocellulose: A Mini-Review on Types and Use in Drug Delivery Systems. Carbohydr. Polym. Technol. Appl. 2021, 2, 100031–100041. [Google Scholar] [CrossRef]
- Abushammala, H.; Mao, J. A Review of the Surface Modification of Cellulose and Nanocellulose Using Aliphatic and Aromatic Mono- and Di-Isocyanates. Molecules 2019, 24, 2782. [Google Scholar] [CrossRef]
- Trache, D.; Hussin, M.H.; Haafiz, M.K.M.; Thakur, V.K. Recent Progress in Cellulose Nanocrystals: Sources and Production. Nanoscale 2017, 9, 1763–1786. [Google Scholar] [CrossRef]
- French, A.D. Glucose, Not Cellobiose, Is the Repeating Unit of Cellulose and Why That Is Important. Cellulose 2017, 24, 4605–4609. [Google Scholar] [CrossRef]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose Nanocrystals: Chemistry, Self-Assembly, and Applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, V.B.; Soetaredjo, F.E.; Putro, J.N.; Santoso, S.P.; Yuliana, M.; Sunarso, J.; Ju, Y.-H.; Ismadji, S. Nanocelluloses: Sources, Pretreatment, Isolations, Modification, and Its Application as the Drug Carriers. Polymers 2021, 13, 2052. [Google Scholar] [CrossRef] [PubMed]
- Díaz, A.; Puiggalí, J. Hydrogels for Biomedical Applications: Cellulose, Chitosan, and Protein/Peptide Derivatives. Gels 2017, 3, 27. [Google Scholar] [CrossRef]
- Grunin, L.Y.; Grunin, Y.B.; Nikolskaya, E.A.; Sheveleva, N.N.; Nikolaev, I.A. An NMR Relaxation and Spin Diffusion Study of Cellulose Structure during Water Adsorption. Biophysics 2017, 62, 198–206. [Google Scholar] [CrossRef]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose Nanomaterials Review: Structure, Properties and Nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef]
- Grunin, Y.B.; Grunin, L.Y.; Schiraya, V.Y.; Ivanova, M.S.; Masas, D.S. Cellulose–Water System’s State Analysis by Proton Nuclear Magnetic Resonance and Sorption Measurements. Bioresour. Bioprocess. 2020, 7, 41–52. [Google Scholar] [CrossRef]
- Smirnova, L.G.; Grunin, Y.B.; Krasil’nikova, S.V.; Zaverkina, M.A.; Bakieva, D.R.; Smirnov, E.V. Study of the Structure and Sorption Properties of Some Types of Cellulose. Colloid J. 2003, 65, 778–781. [Google Scholar] [CrossRef]
- Grunin, L.Y.; Grunin, Y.B.; Talantsev, V.I.; Nikolskaya, E.A.; Masas, D.S. Features of the Structural Organization and Sorption Properties of Cellulose. Polym. Sci. Ser. A 2015, 57, 43–51. [Google Scholar] [CrossRef]
- Li, Q.; Renneckar, S. Supramolecular Structure Characterization of Molecularly Thin Cellulose i Nanoparticles. Biomacromolecules 2011, 12, 650–659. [Google Scholar] [CrossRef]
- Ding, S.Y.; Zhao, S.; Zeng, Y. Size, Shape, and Arrangement of Native Cellulose Fibrils in Maize Cell Walls. Cellulose 2014, 21, 863–871. [Google Scholar] [CrossRef]
- Barhoum, A.; Li, H.; Chen, M.; Cheng, L.; Yang, W.; Dufresne, A. Emerging Applications of Cellulose Nanofibers. In Handbook of Nanofibers; Springer International Publishing: Cham, Switzerland, 2018; pp. 1131–1157. ISBN 9783319536552. [Google Scholar]
- Khalil, H.P.S.A.; Jummaat, F.; Yahya, E.B.; Olaiya, N.G.; Adnan, A.S.; Abdat, M.; Nasir, N.A.M.; Halim, A.S.; Kumar, U.S.U.; Bairwan, R.; et al. A Review on Micro- to Nanocellulose Biopolymer Scaffold Forming for Tissue Engineering Applications. Polymers 2020, 12, 2043. [Google Scholar] [CrossRef] [PubMed]
- Delepierre, G.; Vanderfleet, O.M.; Niinivaara, E.; Zakani, B.; Cranston, E.D. Benchmarking Cellulose Nanocrystals Part II: New Industrially Produced Materials. Langmuir 2021, 37, 8393–8409. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Lyczko, N.; Gopakumar, D.; Maria, H.J.; Nzihou, A.; Thomas, S. Nanocellulose and Its Derivative Materials for Energy and Environmental Applications. J. Mater. Sci. 2022, 57, 6835–6880. [Google Scholar] [CrossRef]
- ISO/TC 229-TS 20477; TAPPI. Standard Terms and Their Definition for Cellulose Nanomaterial. International Organization for Standardization (ISO): Geneva, Swizerland, 2017.
- Yamashita, M.; Yoshida, M.; Matsuo, M.; Sato, S.; Yamamoto, H. Observations of Wood Cell Walls with a Scanning Probe Microscope. Mater. Sci. Appl. 2016, 7, 644–653. [Google Scholar] [CrossRef]
- Athukoralalage, S.S.; Balu, R.; Dutta, N.K.; Roy Choudhury, N. 3D Bioprinted Nanocellulose-Based Hydrogels for Tissue Engineering Applications: A Brief Review. Polymers 2019, 11, 898. [Google Scholar] [CrossRef] [PubMed]
- Phanthong, P.; Reubroycharoen, P.; Hao, X.; Xu, G.; Abudula, A.; Guan, G. Nanocellulose: Extraction and Application. Carbon Resour. Convers. 2018, 1, 32–43. [Google Scholar] [CrossRef]
- Li, N.; Lu, W.; Yu, J.; Xiao, Y.; Liu, S.; Gan, L.; Huang, J. Rod-like Cellulose Nanocrystal/Cis-Aconityl-Doxorubicin Prodrug: A Fluorescence-Visible Drug Delivery System with Enhanced Cellular Uptake and Intracellular Drug Controlled Release. Mater. Sci. Eng. C 2018, 91, 179–189. [Google Scholar] [CrossRef]
- Wang, N.; Ding, E.; Cheng, R. Thermal Degradation Behaviors of Spherical Cellulose Nanocrystals with Sulfate Groups. Polymer 2007, 48, 3486–3493. [Google Scholar] [CrossRef]
- Ait Benhamou, A.; Kassab, Z.; Nadifiyine, M.; Salim, M.H.; Sehaqui, H.; Moubarik, A.; El Achaby, M. Extraction, Characterization and Chemical Functionalization of Phosphorylated Cellulose Derivatives from Giant Reed Plant. Cellulose 2021, 28, 4625–4642. [Google Scholar] [CrossRef]
- Guimaraes, M.; Botara, V.R.; Novack, K.M.; Neto, W.P.F.; Mendes, L.M.; Tonoli, G.H.D. Preparation of Cellulose Nanofibrils from Bamboo Pulp by Mechanical Defibrillation for Their Applications in Biodegradable Composites. J. Nanosci. Nanotechnol. 2015, 15, 6751–6768. [Google Scholar] [CrossRef]
- Benhamou, K.; Kaddami, H.; Magnin, A.; Dufresne, A.; Ahmad, A. Bio-Based Polyurethane Reinforced with Cellulose Nanofibers: A Comprehensive Investigation on the Effect of Interface. Carbohydr. Polym. 2015, 122, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, N.F.; Feitosa, J.P.A.; da Gama, F.M.P.; Morais, J.P.S.; Andrade, F.K.; de Souza, M.D.S.M.; de Freitas Rosa, M. Bacterial Cellulose Nanocrystals Produced under Different Hydrolysis Conditions: Properties and Morphological Features. Carbohydr. Polym. 2017, 155, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhu, C.; Yang, J.; Nie, Y.; Chen, C.; Sun, D. Recent Advances in Bacterial Cellulose. Cellulose 2014, 21, 57–91. [Google Scholar] [CrossRef]
- Gorgieva, S.; Trček, J. Bacterial Cellulose: Production, Modification and Perspectives in Biomedical Applications. Nanomaterials 2019, 9, 1352. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.C.; Yano, H.; Nogi, M.; Eichhorn, S.J. An Estimation of the Young’s Modulus of Bacterial Cellulose Filaments. Cellulose 2008, 15, 507–513. [Google Scholar] [CrossRef]
- Meneguin, A.B.; da Silva Barud, H.; Sábio, R.M.; de Sousa, P.Z.; Manieri, K.F.; de Freitas, L.A.P.; Pacheco, G.; Alonso, J.D.; Chorilli, M. Spray-Dried Bacterial Cellulose Nanofibers: A New Generation of Pharmaceutical Excipient Intended for Intestinal Drug Delivery. Carbohydr. Polym. 2020, 249, 116838–116871. [Google Scholar] [CrossRef] [PubMed]
- Czaja, W.; Romanovicz, D.; Brown, R. malcolm Structural Investigations of Microbial Cellulose Produced in Stationary and Agitated Culture. Cellulose 2004, 11, 403–411. [Google Scholar] [CrossRef]
- Kostić, M. Development of Novel Cellulose-Based Functional Materials. Adv. Technol. 2021, 10, 73–83. [Google Scholar] [CrossRef]
- Ram, B.; Chauhan, G.S. New Spherical Nanocellulose and Thiol-Based Adsorbent for Rapid and Selective Removal of Mercuric Ions. Chem. Eng. J. 2018, 331, 587–596. [Google Scholar] [CrossRef]
- Kim, C.-W.; Kim, D.-S.; Kang, S.-Y.; Marquez, M.; Joo, Y.L. Structural Studies of Electrospun Cellulose Nanofibers. Polymer 2006, 47, 5097–5107. [Google Scholar] [CrossRef]
- Chávez-Guerrero, L.; Sepúlveda-Guzmán, S.; Silva-Mendoza, J.; Aguilar-Flores, C.; Pérez-Camacho, O. Eco-Friendly Isolation of Cellulose Nanoplatelets through Oxidation under Mild Conditions. Carbohydr. Polym. 2018, 181, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.; Pinto, F.; Lourenço, A.F.; Ferreira, P.J.T.; Louro, H.; Silva, M.J. On the Toxicity of Cellulose Nanocrystals and Nanofibrils in Animal and Cellular Models. Cellulose 2020, 27, 5509–5544. [Google Scholar] [CrossRef]
- Roman, M.; Winter, W.T. Effect of Sulfate Groups from Sulfuric Acid Hydrolysis on the Thermal Degradation Behavior of Bacterial Cellulose. Biomacromolecules 2004, 5, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.A.; Foster, E.J. Isolation of Thermally Stable Cellulose Nanocrystals from Spent Coffee Grounds via Phosphoric Acid Hydrolysis. J. Renew. Mater. 2020, 8, 187–203. [Google Scholar] [CrossRef]
- Michelin, M.; Gomes, D.G.; Romaní, A.; Polizeli, M.d.L.T.M.; Teixeira, J.A. Nanocellulose Production: Exploring the Enzymatic Route and Residues of Pulp and Paper Industry. Molecules 2020, 25, 3411. [Google Scholar] [CrossRef] [PubMed]
- Mohd Amin, K.N.; Annamalai, P.K.; Morrow, I.C.; Martin, D. Production of Cellulose Nanocrystals via a Scalable Mechanical Method. RSC Adv. 2015, 5, 57133–57140. [Google Scholar] [CrossRef]
- Saxena, I.M.; Brown, R.M. Cellulose Biosynthesis: Current Views and Evolving Concepts. Ann. Bot. 2005, 96, 9–21. [Google Scholar] [CrossRef]
- Revol, J.F.; Bradford, H.; Giasson, J.; Marchessault, R.H.; Gray, D.G. Helicoidal Self-Ordering of Cellulose Microfibrils in Aqueous Suspension. Int. J. Biol. Macromol. 1992, 14, 170–172. [Google Scholar] [CrossRef]
- Sun, Y.; Lin, L.; Pang, C.; Deng, H.; Peng, H.; Li, J.; He, B.; Liu, S. Hydrolysis of Cotton Fiber Cellulose in Formic Acid. Energy Fuels 2007, 21, 2386–2389. [Google Scholar] [CrossRef]
- Lee, S.Y.; Mohan, D.J.; Kang, I.A.; Doh, G.H.; Lee, S.; Han, S.O. Nanocellulose Reinforced PVA Composite Films: Effects of Acid Treatment and Filler Loading. Fibers Polym. 2009, 10, 77–82. [Google Scholar] [CrossRef]
- Khan, A.; Jawaid, M.; Kian, L.K.; Khan, A.A.P.; Asiri, A.M. Isolation and Production of Nanocrystalline Cellulose from Conocarpus Fiber. Polymers 2021, 13, 1835. [Google Scholar] [CrossRef] [PubMed]
- Oprea, S.; Potolinca, V.O.; Oprea, V. Physical Properties and the Ability to Disperse into Different Polar Solvents of the New Polyurethane–Cellulose Composites. J. Elastomers Plast. 2020, 52, 548–572. [Google Scholar] [CrossRef]
- Khadivi, P.; Salami-Kalajahi, M.; Roghani-Mamaqani, H.; Lotfi Mayan Sofla, R. Fabrication of Microphase-Separated Polyurethane/Cellulose Nanocrystal Nanocomposites with Irregular Mechanical and Shape Memory Properties. Appl. Phys. A 2019, 125, 779–789. [Google Scholar] [CrossRef]
- Amin, K.N.M.; Annamalai, P.K.; Martin, D. Cellulose Nanocrystals with Enhanced Thermal Stability Reinforced Thermoplastic Polyurethane. Malays. J. Anal. Sci. 2017, 21, 754–761. [Google Scholar] [CrossRef]
- Achor, M.; Oyeniyi, Y.J.; Yahaya, A. Extraction and Characterization of Microcrystalline Cellulose Obtained from the Back of the Fruit of Lageriana Siceraria (Water Gourd). J. Appl. Pharm. Sci. 2014, 4, 57–60. [Google Scholar] [CrossRef]
- Ou, H.; Chen, G.; Zhu, P.; Wei, Y.; Li, F. Preparation and Strain Sensitive Performance of Cellulose Nanofiber-Carbon Nanotubes/Thermoplastic Polyurethane Composite Films. Acta Mater. Compos. Sin. 2020, 37, 2735–2742. [Google Scholar] [CrossRef]
- Larraza, I.; Vadillo, J.; Santamaria-Echart, A.; Tejado, A.; Azpeitia, M.; Vesga, E.; Orue, A.; Saralegi, A.; Arbelaiz, A.; Eceiza, A. The Effect of the Carboxylation Degree on Cellulose Nanofibers and Waterborne Polyurethane/Cellulose Nanofiber Nanocomposites Properties. Polym. Degrad. Stab. 2020, 173, 109084–109096. [Google Scholar] [CrossRef]
- Lasseuguette, E.; Roux, D.; Nishiyama, Y. Rheological Properties of Microfibrillar Suspension of TEMPO-Oxidized Pulp. Cellulose 2008, 15, 425–433. [Google Scholar] [CrossRef]
- Barja, F. Bacterial Nanocellulose Production and Biomedical Applications. J. Biomed. Res. 2021, 35, 310–317. [Google Scholar] [CrossRef]
- Verpaalen, R.C.P.; Engels, T.; Schenning, A.P.H.J.; Debije, M.G. Stimuli-Responsive Shape Changing Commodity Polymer Composites and Bilayers. ACS Appl. Mater. Interfaces 2020, 12, 38829–38844. [Google Scholar] [CrossRef]
- Haskew, M.J.; Hardy, J.G. A Mini-Review of Shape-Memory Polymer-Based Materials: Stimuli-Responsive Shape-Memory Polymers. Johns. Matthey Technol. Rev. 2020, 64, 425–442. [Google Scholar] [CrossRef]
- Behl, M.; Razzaq, M.Y.; Lendlein, A. Multifunctional Shape-Memory Polymers. Adv. Mater. 2010, 22, 3388–3410. [Google Scholar] [CrossRef] [PubMed]
- Piegat, A.; El Fray, M. Thermoplastic Elastomers: Materials for Heart Assist Devices. Polym. Med. 2016, 46, 79–87. [Google Scholar] [CrossRef]
- Gupta, A.; Maharjan, A.; Kim, B.S. Shape Memory Polyurethane and Its Composites for Various Applications. Appl. Sci. 2019, 9, 4694. [Google Scholar] [CrossRef]
- Naureen, B.; Haseeb, A.S.M.A.; Basirun, W.J.; Muhamad, F. Recent Advances in Tissue Engineering Scaffolds Based on Polyurethane and Modified Polyurethane. Mater. Sci. Eng. C 2021, 118, 111228–111232. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, M.A.; Anokhin, D.V.; Badamshina, E.R. Recent Advances in the Synthesis and Application of Thermoplastic Semicrystalline Shape Memory Polyurethanes. Polym. Sci. Ser. B 2020, 62, 427–450. [Google Scholar] [CrossRef]
- Fisher, H.; Woolard, P.; Ross, C.; Kunkel, R.; Bohnstedt, B.N.; Liu, Y.; Lee, C.-H. Thermomechanical Data of Polyurethane Shape Memory Polymer: Considering Varying Compositions. Data Brief 2020, 32, 106294–106304. [Google Scholar] [CrossRef]
- Bratasyuk, N.A.; Zuev, V.V. The Effect Molecular Weight of Polyol Components on Shape Memory Effect of Epoxy-polyurethane Composites. Polym. Eng. Sci. 2021, 61, 2674–2690. [Google Scholar] [CrossRef]
- Kumar, B.; Noor, N.; Thakur, S.; Pan, N.; Narayana, H.; Yan, S.; Wang, F.; Shah, P. Shape Memory Polyurethane-Based Smart Polymer Substrates for Physiologically Responsive, Dynamic Pressure (Re)Distribution. ACS Omega 2019, 4, 15348–15358. [Google Scholar] [CrossRef]
- Mirtschin, N.; Pretsch, T. Designing Temperature-Memory Effects in Semicrystalline Polyurethane. RSC Adv. 2015, 5, 46307–46315. [Google Scholar] [CrossRef]
- Schönfeld, D.; Chalissery, D.; Wenz, F.; Specht, M.; Eberl, C.; Pretsch, T. Actuating Shape Memory Polymer for Thermoresponsive Soft Robotic Gripper and Programmable Materials. Molecules 2021, 26, 522. [Google Scholar] [CrossRef]
- Jin, X.; Guo, N.; You, Z.; Tan, Y. Design and Performance of Polyurethane Elastomers Composed with Different Soft Segments. Materials 2020, 13, 4991. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kang, S.-K. Principles for Controlling the Shape Recovery and Degradation Behavior of Biodegradable Shape-Memory Polymers in Biomedical Applications. Micromachines 2021, 12, 757. [Google Scholar] [CrossRef]
- Anokhin, D.V.; Gorbunova, M.A.; Abukaev, A.F.; Ivanov, D.A. Multiblock Thermoplastic Polyurethanes: In-situ Studies of Structural and Morphological Evolution under Strain. Materials 2021, 14, 3009. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.-I.; Park, H.-S.; Park, J.-H.; Knowles, J.C.; Gong, M.-S. Application of High-Strength Biodegradable Polyurethanes Containing Different Ratios of Biobased Isomannide and Poly (ϵ-Caprolactone) Diol. J. Bioact. Compat. Polym. 2013, 28, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Król, P.; Uram, Ł.; Król, B.; Pielichowska, K.; Sochacka-Piętal, M.; Walczak, M. Synthesis and Property of Polyurethane Elastomer for Biomedical Applications Based on Nonaromatic Isocyanates, Polyesters, and Ethylene Glycol. Colloid Polym. Sci. 2020, 298, 1077–1093. [Google Scholar] [CrossRef]
- Uscátegui, Y.L.; Arévalo-Alquichire, S.J.; Gómez-Tejedor, J.A.; Vallés-Lluch, A.; Díaz, L.E.; Valero, M.F. Polyurethane-Based Bioadhesive Synthesized from Polyols Derived from Castor Oil (Ricinus communis) and Low Concentration of Chitosan. J. Mater. Res. 2017, 32, 3699–3711. [Google Scholar] [CrossRef]
- Sankar, G.; Yan, N. Synthesis and Deblocking Studies of Low Temperature Heat-Curable Blocked Polymeric Methylene Diphenyl Diisocyanates. J. Macromol. Sci. Part A Pure Appl. Chem. 2015, 52, 47–55. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Yang, G.; Zheng, X.; Zhou, S. Multi-Stimulus-Responsive Shape-Memory Polymer Nanocomposite Network Cross-Linked by Cellulose Nanocrystals. ACS Appl. Mater. Interfaces 2015, 7, 4118–4126. [Google Scholar] [CrossRef]
- Raschip, I.E.; Moldovan, L.; Stefan, L.; Oancea, A.; Vasile, C. Compatibility and Cytotoxicity Testing of Some New Biomaterials Based on Polyurethane and Hydroxypropylcellulose Blends. Optoelectron. Adv. Mater.-Rapid Commun. 2009, 3, 1336–1342. [Google Scholar]
- Karpov, S.V.; Dzhalmukhanova, A.S.; Chernyayev, D.A.; Lodygina, V.P.; Firsova, A.I.; Badamshina, E.R. The Investigation of Triethylammonium Carboxylates Influence on the Kinetics of Urethane Formation Processing during Waterborne Polyurethane Synthesis. Polym. Adv. Technol. 2021, 32, 2727–2734. [Google Scholar] [CrossRef]
- Karpov, S.V.; Lodygina, V.P.; Komratova, V.V.; Dzhalmukhanova, A.S.; Malkov, G.V.; Badamshina, E.R. Kinetics of Urethane Formation from Isophorone Diisocyanate: The Catalyst and Solvent Effects. Kinet. Catal. 2016, 57, 422–428. [Google Scholar] [CrossRef]
- Singh, A.; Kumar, R.; Soni, P.K.; Singh, V. Investigation of the Effect of Diisocyanate on the Thermal Degradation Behavior and Degradation Kinetics of Polyether-Based Polyurethanes. J. Macromol. Sci. Part B Phys. 2020, 59, 775–795. [Google Scholar] [CrossRef]
- Javaid, M.A.; Zia, K.M.; Khera, R.A.; Jabeen, S.; Mumtaz, I.; Younis, M.A.; Shoaib, M.; Bhatti, I.A. Evaluation of Cytotoxicity, Hemocompatibility and Spectral Studies of Chitosan Assisted Polyurethanes Prepared with Various Diisocyanates. Int. J. Biol. Macromol. 2019, 129, 116–126. [Google Scholar] [CrossRef]
- Uscátegui, Y.L.; Díaz, L.E.; Valero, M.F. In Vitro and in Vivo Biocompatibility of Polyurethanes Synthesized with Castor Oil Polyols for Biomedical Devices. J. Mater. Res. 2019, 34, 519–531. [Google Scholar] [CrossRef]
- Krol, P. Synthesis Methods, Chemical Structures and Phase Structures of Linear Polyurethanes. Properties and Applications of Linear Polyurethanes in Polyurethane Elastomers, Copolymers and Ionomers. Prog. Mater. Sci. 2007, 52, 915–1015. [Google Scholar] [CrossRef]
- Anokhin, D.V.; Gorbunova, M.A.; Estrin, Y.I.; Komratova, V.V.; Badamshina, E.R. The Role of Fast and Slow Processes in the Formation of Structure and Properties of Thermoplastic Polyurethanes. Phys. Chem. Chem. Phys. 2016, 18, 31769–31776. [Google Scholar] [CrossRef]
- Gorbunova, M.A.; Shukhardin, D.M.; Lesnichaya, V.A.; Badamshina, E.R.; Anokhin, D.V. New Polyurethane Urea Thermoplastic Elastomers with Controlled Mechanical and Thermal Properties for Medical Applications. Key Eng. Mater. 2019, 816, 187–191. [Google Scholar] [CrossRef]
- Li, L.; Xu, L.; Ding, W.; Lu, H.; Zhang, C.; Liu, T. Molecular-Engineered Hybrid Carbon Nanofillers for Thermoplastic Polyurethane Nanocomposites with High Mechanical Strength and Toughness. Compos. Part B Eng. 2019, 177, 107381–107391. [Google Scholar] [CrossRef]
- Yao, Z.; Wu, D.; Chen, C.; Zhang, M. Creep Behavior of Polyurethane Nanocomposites with Carbon Nanotubes. Compos. Part A Appl. Sci. Manuf. 2013, 50, 65–72. [Google Scholar] [CrossRef]
- Marcovich, N.E.; Auad, M.L.; Bellesi, N.E.; Nutt, S.R.; Aranguren, M.I. Cellulose Micro/Nanocrystals Reinforced Polyurethane. J. Mater. Res. 2006, 21, 870–881. [Google Scholar] [CrossRef]
- Bras, J.; Viet, D.; Bruzzese, C.; Dufresne, A. Correlation between Stiffness of Sheets Prepared from Cellulose Whiskers and Nanoparticles Dimensions. Carbohydr. Polym. 2011, 84, 211–215. [Google Scholar] [CrossRef]
- Iwamoto, S.; Nakagaito, A.N.; Yano, H. Nano-Fibrillation of Pulp Fibers for the Processing of Transparent Nanocomposites. Appl. Phys. A 2007, 89, 461–466. [Google Scholar] [CrossRef]
- Nogi, M.; Iwamoto, S.; Nakagaito, A.N.; Yano, H. Optically Transparent Nanofiber Paper. Adv. Mater. 2009, 21, 1595–1598. [Google Scholar] [CrossRef]
- Pei, A.; Malho, J.M.; Ruokolainen, J.; Zhou, Q.; Berglund, L.A. Strong Nanocomposite Reinforcement Effects in Polyurethane Elastomer with Low Volume Fraction of Cellulose Nanocrystals. Macromolecules 2011, 44, 4422–4427. [Google Scholar] [CrossRef]
- Prataviera, R.; Pollet, E.; Bretas, R.E.S.; Avérous, L.; de Almeida Lucas, A. Melt Processing of Nanocomposites of Cellulose Nanocrystals with Biobased Thermoplastic Polyurethane. J. Appl. Polym. Sci. 2021, 138, 12–20. [Google Scholar] [CrossRef]
- Noormohammadi, F.; Nourany, M.; Mir Mohamad Sadeghi, G.; Wang, P.-Y.; Shahsavarani, H. The Role of Cellulose Nanowhiskers in Controlling Phase Segregation, Crystallization and Thermal Stimuli Responsiveness in PCL-PEGx-PCL Block Copolymer-Based PU for Human Tissue Engineering Applications. Carbohydr. Polym. 2021, 252, 117219–117227. [Google Scholar] [CrossRef]
- Du, W.; Zhang, Z.; Yin, C.; Ge, X.; Shi, L. Preparation of Shape Memory Polyurethane/Modified Cellulose Nanocrystals Composites with Balanced Comprehensive Performances. Polym. Adv. Technol. 2021, 32, 4710–4720. [Google Scholar] [CrossRef]
- Bi, H.; Ren, Z.; Ye, G.; Sun, H.; Guo, R.; Jia, X.; Xu, M. Fabrication of Cellulose Nanocrystal Reinforced Thermoplastic Polyurethane/Polycaprolactone Blends for Three-Dimension Printing Self-Healing Nanocomposites. Cellulose 2020, 27, 8011–8026. [Google Scholar] [CrossRef]
- Nicharat, A.; Shirole, A.; Foster, E.J.; Weder, C. Thermally Activated Shape Memory Behavior of Melt-Mixed Polyurethane/Cellulose Nanocrystal Composites. J. Appl. Polym. Sci. 2017, 134, 45033–45043. [Google Scholar] [CrossRef]
- Mabrouk, A.B.; Kaddami, H.; Boufi, S.; Erchiqui, F.; Dufresne, A. Cellulosic Nanoparticles from Alfa Fibers (Stipa Tenacissima): Extraction Procedures and Reinforcement Potential in Polymer Nanocomposites. Cellulose 2012, 19, 843–853. [Google Scholar] [CrossRef]
- Bendahou, A.; Kaddami, H.; Dufresne, A. Investigation on the Effect of Cellulosic Nanoparticles’ Morphology on the Properties of Natural Rubber Based Nanocomposites. Eur. Polym. J. 2010, 46, 609–620. [Google Scholar] [CrossRef]
- Pandey, J.K.; Lee, C.S.; Ahn, S.-H. Preparation and Properties of Bio-Nanoreinforced Composites from Biodegradable Polymer Matrix and Cellulose Whiskers. J. Appl. Polym. Sci. 2010, 115, 2493–2501. [Google Scholar] [CrossRef]
- Eyley, S.; Thielemans, W. Surface Modification of Cellulose Nanocrystals. Nanoscale 2014, 6, 7764–7779. [Google Scholar] [CrossRef]
- Shi, Z.; Li, S.; Li, M.; Gan, L.; Huang, J. Surface Modification of Cellulose Nanocrystals towards New Materials Development. J. Appl. Polym. Sci. 2021, 138, e51555–e51576. [Google Scholar] [CrossRef]
- Shirole, A.; Nicharat, A.; Perotto, C.U.; Weder, C. Tailoring the Properties of a Shape-Memory Polyurethane via Nanocomposite Formation and Nucleation. Macromolecules 2018, 51, 1841–1849. [Google Scholar] [CrossRef]
- Cai, C.; Wei, Z.; Wang, X.; Mei, C.; Fu, Y.; Zhong, W.H. Novel Double-Networked Polyurethane Composites with Multi-Stimuli Responsive Functionalities. J. Mater. Chem. A 2018, 6, 17457–17472. [Google Scholar] [CrossRef]
- Rueda, L.; Fernández d’Arlas, B.; Zhou, Q.; Berglund, L.A.; Corcuera, M.A.; Mondragon, I.; Eceiza, A. Isocyanate-Rich Cellulose Nanocrystals and Their Selective Insertion in Elastomeric Polyurethane. Compos. Sci. Technol. 2011, 71, 1953–1960. [Google Scholar] [CrossRef]
- Tian, D.; Wang, F.; Yang, Z.; Niu, X.; Wu, Q.; Sun, P. High-Performance Polyurethane Nanocomposites Based on UPy-Modified Cellulose Nanocrystals. Carbohydr. Polym. 2019, 219, 191–200. [Google Scholar] [CrossRef]
- Girouard, N.M.; Xu, S.; Schueneman, G.T.; Shofner, M.L.; Meredith, J.C. Site-Selective Modification of Cellulose Nanocrystals with Isophorone Diisocyanate and Formation of Polyurethane-CNC Composites. ACS Appl. Mater. Interfaces 2016, 8, 1458–1467. [Google Scholar] [CrossRef]
- Abushammala, H.; Mao, J. Impact of the Surface Properties of Cellulose Nanocrystals on the Crystallization Kinetics of Poly(Butylene Succinate). Crystals 2020, 10, 196. [Google Scholar] [CrossRef]
- Abushammala, H. Nano-Brushes of Alcohols Grafted onto Cellulose Nanocrystals for Reinforcing Poly(Butylene Succinate): Impact of Alcohol Chain Length on Interfacial Adhesion. Polymers 2020, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Abushammala, H. On the Para/Ortho Reactivity of Isocyanate Groups during the Carbamation of Cellulose Nanocrystals Using 2,4-Toluene Diisocyanate. Polymers 2019, 11, 1164. [Google Scholar] [CrossRef]
- Abushammala, H. A Simple Method for the Quantification of Free Isocyanates on the Surface of Cellulose Nanocrystals upon Carbamation Using Toluene Diisocyanate. Surfaces 2019, 2, 444–454. [Google Scholar] [CrossRef]
- Gwon, J.G.; Cho, H.J.; Chun, S.J.; Lee, S.; Wu, Q.; Li, M.C.; Lee, S.Y. Mechanical and Thermal Properties of Toluene Diisocyanate-Modified Cellulose Nanocrystal Nanocomposites Using Semi-Crystalline Poly(Lactic Acid) as a Base Matrix. RSC Adv. 2016, 6, 73879–73886. [Google Scholar] [CrossRef]
- Semsarzadeh, M.A.; Navarchian, A.H. Kinetic Study of the Bulk Reaction between Tdi and Ppg in Presence of Dbtdl and Feaa Catalysts Using Quantitative Ftir Spectroscopy. J. Polym. Eng. 2003, 23, 225–240. [Google Scholar] [CrossRef]
- Zaverkina, M.A.; Lodygina, V.P.; Komratova, V.V.; Stovbun, E.V.; Badamshina, E.R. Kinetics of Diisocyanate Reactions with Chain-Extending Agents. Polym. Sci. Ser. A 2006, 48, 382–387. [Google Scholar] [CrossRef]
- Špirková, M.; Kubín, M.; Dušek, K. Side Reactions in the Formation of Polyurethanes: Model Reactions Between Phenylisocyanate and 1-Butanol. J. Macromol. Sci. Part A-Chem. 1987, 24, 1151–1166. [Google Scholar] [CrossRef]
- Morandi, G.; Thielemans, W. Synthesis of Cellulose Nanocrystals Bearing Photocleavable Grafts by ATRP. Polym. Chem. 2012, 3, 1402–1407. [Google Scholar] [CrossRef]
- Karpov, S.V.; Lodygina, V.P.; Komratova, V.V.; Dzhalmukhanova, A.S.; Malkov, G.V.; Badamshina, E.R. Kinetics of Urethane Formation from Isophorone Diisocyanate: The Alcohol Nature Effect. Kinet. Catal. 2016, 57, 319–325. [Google Scholar] [CrossRef]
- Lomölder, R.; Plogmann, F.; Speier, P. Selectivity of Isophorone Diisocyanate in the Urethane Reaction Influence of Temperature, Catalysis, and Reaction Partners. J. Coat. Technol. 1997, 69, 51–57. [Google Scholar] [CrossRef]
- De Mesquita, J.P.; Donnici, C.L.; Pereira, F.V. Biobased Nanocomposites from Layer-by-Layer Assembly of Cellulose Nanowhiskers with Chitosan. Biomacromolecules 2010, 11, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Long, L.-Y.; Weng, Y.-X.; Wang, Y.-Z. Cellulose Aerogels: Synthesis, Applications, and Prospects. Polymers 2018, 10, 623. [Google Scholar] [CrossRef] [PubMed]
- Capadona, J.R.; Van Den Berg, O.; Capadona, L.A.; Schroeter, M.; Rowan, S.J.; Tyler, D.J.; Weder, C. A Versatile Approach for the Processing of Polymer Nanocomposites with Self-Assembled Nanofibre Templates. Nat. Nanotechnol. 2007, 2, 765–769. [Google Scholar] [CrossRef]
- Shi, Z.; Huang, J.; Liu, C.; Ding, B.; Kuga, S.; Cai, J.; Zhang, L. Three-Dimensional Nanoporous Cellulose Gels as a Flexible Reinforcement Matrix for Polymer Nanocomposites. ACS Appl. Mater. Int. 2015, 7, 22990–22998. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wei, P.; Huang, J.; Xu, D.; Zhong, Y.; Hu, L.; Zhang, L.; Cai, J. Mechanically Strong Shape-Memory and Solvent-Resistant Double-Network Polyurethane/Nanoporous Cellulose Gel Nanocomposites. ACS Sustain. Chem. Eng. 2019, 7, 15974–15982. [Google Scholar] [CrossRef]
- Amin, K.N.M.; Amiralian, N.; Annamalai, P.K.; Edwards, G.; Chaleat, C.; Martin, D.J. Scalable Processing of Thermoplastic Polyurethane Nanocomposites Toughened with Nanocellulose. Chem. Eng. J. 2016, 302, 406–416. [Google Scholar] [CrossRef]
- Seydibeyoǧlu, M.Ö.; Misra, M.; Mohanty, A.; Blaker, J.J.; Lee, K.Y.; Bismarck, A.; Kazemizadeh, M. Green Polyurethane Nanocomposites from Soy Polyol and Bacterial Cellulose. J. Mater. Sci. 2013, 48, 2167–2175. [Google Scholar] [CrossRef]
- Özgür Seydibeyoǧlu, M.; Oksman, K. Novel Nanocomposites Based on Polyurethane and Micro Fibrillated Cellulose. Compos. Sci. Technol. 2008, 68, 908–914. [Google Scholar] [CrossRef]
- Khadivi, P.; Salami-Kalajahi, M.; Roghani-Mamaqani, H.; Sofla, R.L.M. Polydimethylsiloxane-based Polyurethane/Cellulose Nanocrystal Nanocomposites: From Structural Properties Toward Cytotoxicity. Silicon 2021, 14, 1695–1703. [Google Scholar] [CrossRef]
- Saralegi, A.; Gonzalez, M.L.; Valea, A.; Eceiza, A.; Corcuera, M.A. The Role of Cellulose Nanocrystals in the Improvement of the Shape-Memory Properties of Castor Oil-Based Segmented Thermoplastic Polyurethanes. Compos. Sci. Technol. 2014, 92, 27–33. [Google Scholar] [CrossRef]
- Lee, M.; Heo, M.H.; Lee, H.H.; Kim, Y.W.; Shin, J. Tunable Softening and Toughening of Individualized Cellulose Nanofibers-Polyurethane Urea Elastomer Composites. Carbohydr. Polym. 2017, 159, 125–135. [Google Scholar] [CrossRef]
- Rueda, L.; Saralegi, A.; Fernández-d’Arlas, B.; Zhou, Q.; Alonso-Varona, A.; Berglund, L.A.; Mondragon, I.; Corcuera, M.A.; Eceiza, A. In-situ Polymerization and Characterization of Elastomeric Polyurethane-Cellulose Nanocrystal Nanocomposites. Cell Response Evaluation. Cellulose 2013, 20, 1819–1828. [Google Scholar] [CrossRef]
- Rueda, L.; Saralegui, A.; Fernández D’Arlas, B.; Zhou, Q.; Berglund, L.A.; Corcuera, M.A.; Mondragon, I.; Eceiza, A. Cellulose Nanocrystals/Polyurethane Nanocomposites. Study from the Viewpoint of Microphase Separated Structure. Carbohydr. Polym. 2013, 92, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Auad, M.L.; Mosiewicki, M.A.; Richardson, T.; Aranguren, M.I.; Marcovich, N.E. Nanocomposites Made from Cellulose Nanocrystals and Tailored Segmented Polyurethanes. J. Appl. Polym. Sci. 2010, 115, 1215–1225. [Google Scholar] [CrossRef]
- Shahrousvand, E.; Shahrousvand, M. Preparation of Polyurethane/Poly (2-Hydroxyethyl Methacrylate) Semi-IPNs Containing Cellulose Nanocrystals for Biomedical Applications. Mater. Today Commun. 2021, 27, 102421–102432. [Google Scholar] [CrossRef]
- Shahrousvand, M.; Ghollasi, M.; Zarchi, A.A.K.; Salimi, A. Osteogenic Differentiation of HMSCs on Semi-Interpenetrating Polymer Networks of Polyurethane/Poly(2-hydroxyethyl Methacrylate)/Cellulose Nanowhisker Scaffolds. Int. J. Biol. Macromol. 2019, 138, 262–271. [Google Scholar] [CrossRef]
- Saralegi, A.; Rueda, L.; Martin, L.; Arbelaiz, A.; Eceiza, A.; Corcuera, M.A. From Elastomeric to Rigid Polyurethane/Cellulose Nanocrystal Bionanocomposites. Compos. Sci. Technol. 2013, 88, 39–47. [Google Scholar] [CrossRef]
- Brakat, A.; Zhu, H. Nanocellulose-Graphene Hybrids: Advanced Functional Materials as Multifunctional Sensing Platform. Nano-Micro Lett. 2021, 13, 94–131. [Google Scholar] [CrossRef]
- Liu, H.; Song, J.; Shang, S.; Song, Z.; Wang, D. Cellulose Nanocrystal/Silver Nanoparticle Composites as Bifunctional Nanofillers within Waterborne Polyurethane. ACS Appl. Mater. Interfaces 2012, 4, 2413–2419. [Google Scholar] [CrossRef]
- Wong, B.S.; Yoong, S.L.; Jagusiak, A.; Panczyk, T.; Ho, H.K.; Ang, W.H.; Pastorin, G. Carbon Nanotubes for Delivery of Small Molecule Drugs. Adv. Drug Deliv. Rev. 2013, 65, 1964–2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Anoshkin, I.V.; Nasibulin, A.G.; Korhonen, J.T.; Seitsonen, J.; Pere, J.; Kauppinen, E.I.; Ras, R.H.A.; Ikkala, O. Modifying Native Nanocellulose Aerogels with Carbon Nanotubes for Mechanoresponsive Conductivity and Pressure Sensing. Adv. Mater. 2013, 25, 2428–2432. [Google Scholar] [CrossRef] [PubMed]
- Olivier, C.; Moreau, C.; Bertoncini, P.; Bizot, H.; Chauvet, O.; Cathala, B. Cellulose Nanocrystal-Assisted Dispersion of Luminescent Single-Walled Carbon Nanotubes for Layer-by-Layer Assembled Hybrid Thin Films. Langmuir 2012, 28, 12463–12471. [Google Scholar] [CrossRef] [PubMed]
- Hamedi, M.M.; Hajian, A.; Fall, A.B.; Hkansson, K.; Salajkova, M.; Lundell, F.; Wgberg, L.; Berglund, L.A. Highly Conducting, Strong Nanocomposites Based on Nanocellulose-Assisted Aqueous Dispersions of Single-Wall Carbon Nanotubes. ACS Nano 2014, 8, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.-S.; Zeng, H.-X.; Wu, J.; Dong, L.-Y.; Zhu, J.-T.; Xue, Z.-G.; Zhou, X.-P.; Xie, X.-L.; Mai, Y.-W. Biocompatible Reduced Graphene Oxide Sheets with Superior Water Dispersibility Stabilized by Cellulose Nanocrystals and Their Polyethylene Oxide Composites. Green Chem. 2016, 18, 1674–1683. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, H.; He, X.; Hu, Y.; Zhu, E.; Gao, Y.; Liu, D.; Shi, Z.; Li, J.; Yang, Q.; et al. Flexible Electronic Skin Sensor Based on Regenerated Cellulose/Carbon Nanotube Composite Films. Cellulose 2020, 27, 10199–10211. [Google Scholar] [CrossRef]
- Bai, Y.; Jiang, C.; Wang, Q.; Wang, T. Multi-Shape-Memory Property Study of Novel Poly(ε-Caprolactone)/Ethyl Cellulose Polymer Networks. Macromol. Chem. Phys. 2013, 214, 2465–2472. [Google Scholar] [CrossRef]
- Wang, W.; Liu, D.; Lu, L.; Chen, H.; Gong, T.; Lv, J.; Zhou, S. The Improvement of the Shape Memory Function of Poly(ϵ-Caprolactone)/Nano-Crystalline Cellulose Nanocomposites via Recrystallization under a High-Pressure Environment. J. Mater. Chem. A 2016, 4, 5984–5992. [Google Scholar] [CrossRef]
- Du, Y.; Li, D.; Liu, L.; Gai, G. Recent Achievements of Self-Healing Graphene/Polymer Composites. Polymers 2018, 10, 114. [Google Scholar] [CrossRef]
- Wu, T.; Frydrych, M.; O’Kelly, K.; Chen, B. Poly(Glycerol Sebacate Urethane)–Cellulose Nanocomposites with Water-Active Shape-Memory Effects. Biomacromolecules 2014, 15, 2663–2671. [Google Scholar] [CrossRef]
- Capadona, J.R.; Shanmuganathan, K.; Tyler, D.J.; Rowan, S.J.; Weder, C. Stimuli-Responsive Polymer Nanocomposites Inspired by the Sea Cucumber Dermis. Science 2008, 319, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yang, G.; Jing, M.; Fu, Q.; Chiu, F.C. Microfibrillated Cellulose-Reinforced Bio-Based Poly(Propylene Carbonate) with Dual Shape Memory and Self-Healing Properties. J. Mater. Chem. A 2014, 2, 20393–20401. [Google Scholar] [CrossRef]
- Tan, L.; Hu, J.; Ying Rena, K.; Zhu, Y.; Liu, P. Quick Water-Responsive Shape Memory Hybrids with Cellulose Nanofibers. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 767–775. [Google Scholar] [CrossRef]
- Zhu, Y.; Hu, J.; Luo, H.; Young, R.J.; Deng, L.; Zhang, S.; Fan, Y.; Ye, G. Rapidly Switchable Water-Sensitive Shape-Memory Cellulose/Elastomer Nano-Composites. Soft Matter 2012, 8, 2509. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, Z.; Liu, Z.; Kang, H.; Liu, Y. Cellulose Nanofibers/Polyurethane Shape Memory Composites with Fast Water-Responsivity. J. Mater. Chem. B 2018, 6, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Hu, J.; Zhu, Y. Polymeric Shape Memory Nanocomposites with Heterogeneous Twin Switches. Macromol. Chem. Phys. 2011, 212, 1981–1986. [Google Scholar] [CrossRef]
- Cao, X.; Xu, C.; Wang, Y.; Liu, Y.; Liu, Y.; Chen, Y. New Nanocomposite Materials Reinforced with Cellulose Nanocrystals in Nitrile Rubber. Polym. Test. 2013, 32, 819–826. [Google Scholar] [CrossRef]
- Ghosal, K.; Agatemor, C.; Tucker, N.; Kny, E.; Thomas, S. Chapter 1. Electrical Spinning to Electrospinning: A Brief History. In Electrospinning: From Basic Research to Commercialization; The Royal Society of Chemistry: London, UK, 2018; pp. 1–23. ISBN 978-1-78801-100-6. [Google Scholar]
- Fleige, E.; Quadir, M.A.; Haag, R. Stimuli-Responsive Polymeric Nanocarriers for the Controlled Transport of Active Compounds: Concepts and Applications. Adv. Drug Deliv. Rev. 2012, 64, 866–884. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Chitins and Chitosans for the Repair of Wounded Skin, Nerve, Cartilage and Bone. Carbohydr. Polym. 2009, 76, 167–182. [Google Scholar] [CrossRef]
- Araki, J.; Wada, M.; Kuga, S.; Okano, T. Flow Properties of Microcrystalline Cellulose Suspension Prepared by Acid Treatment of Native Cellulose. Colloids Surf. A Physicochem. Eng. Asp. 1998, 142, 75–82. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Liu, Y.; Gong, T.; Wang, L.; Zhou, S. Highly PH-Sensitive Polyurethane Exhibiting Shape Memory and Drug Release. Polym. Chem. 2014, 5, 5168–5174. [Google Scholar] [CrossRef]
- Way, A.E.; Hsu, L.; Shanmuganathan, K.; Weder, C.; Rowan, S.J. PH-Responsive Cellulose Nanocrystal Gels and Nanocomposites. ACS Macro Lett. 2012, 1, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz Memiş, N.; Kaplan, S. Dual Responsive Wool Fabric by Cellulose Nanowhisker Reinforced Shape Memory Polyurethane. J. Appl. Polym. Sci. 2020, 137, 28–38. [Google Scholar] [CrossRef]
- Sukhkhawuttigit, S.; Ummartyotin, S.; Infahsaeng, Y. Development of Chemically Grafted Multiwall Carbon Nanotube onto Cellulose Fiber Sheet and Polyurethane Based Resin Composite for an Active Paper. J. Met. Mater. Miner. 2021, 31, 110–117. [Google Scholar] [CrossRef]
- Shrestha, S.; Shrestha, B.K.; Lee, J.; Joong, O.K.; Kim, B.S.; Park, C.H.; Kim, C.S. A Conducting Neural Interface of Polyurethane/Silk-Functionalized Multiwall Carbon Nanotubes with Enhanced Mechanical Strength for Neuroregeneration. Mater. Sci. Eng. C 2019, 102, 511–523. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical structure of cellulose representing reducing and non-reducing ends (a); D-glucose residues (b), the structure of cellulose macrofibrils (c), and variant cross section of macrofibrils consisting of aggregates microfibrils, each containing four elementary fibrils (d). Adapted from [40,43,44].
Figure 1.
Chemical structure of cellulose representing reducing and non-reducing ends (a); D-glucose residues (b), the structure of cellulose macrofibrils (c), and variant cross section of macrofibrils consisting of aggregates microfibrils, each containing four elementary fibrils (d). Adapted from [40,43,44].

Figure 2.
Number of published articles on nanocrystalline cellulose per year from 2002 to 2022 according to Scopus (keywords: «cellulose nanocrystals», «cellulose nanowhiskers», and «nanocrystalline cellulose»).
Figure 2.
Number of published articles on nanocrystalline cellulose per year from 2002 to 2022 according to Scopus (keywords: «cellulose nanocrystals», «cellulose nanowhiskers», and «nanocrystalline cellulose»).

Figure 3.
Hierarchical structure of cellulose. Adapted from [57].
Figure 3.
Hierarchical structure of cellulose. Adapted from [57].

Figure 4.
General scheme of the studied TPUs synthesis, where (a) synthesis, (b) microphase separation structure of hard and soft segments, and (c) structure of typical diisocyanates reagents.
Figure 4.
General scheme of the studied TPUs synthesis, where (a) synthesis, (b) microphase separation structure of hard and soft segments, and (c) structure of typical diisocyanates reagents.

Figure 5.
Producing TPU composites containing nanocellulose.

Figure 6.
Proposed reactive mechanism and structure of double-networked TPU composites (a), shape change pattern of thermo-sensitive TPU composite (b), shape change of TPU composite (c), SEM micrographs of TPU composite before and after thermo-mechanical testing (d), mechanical properties of neat TPU and TPU composites (e). Adapted from [139].
Figure 6.
Proposed reactive mechanism and structure of double-networked TPU composites (a), shape change pattern of thermo-sensitive TPU composite (b), shape change of TPU composite (c), SEM micrographs of TPU composite before and after thermo-mechanical testing (d), mechanical properties of neat TPU and TPU composites (e). Adapted from [139].

Figure 7.
Structure of modified nanocellulose (CNC–UPy) by in situ polymerization (a), AFM images CNC–UPy (b), mechanical properties of native TPU and composites in the presence of modified TPU/CNC−UPy and unmodified TPU/CNC nanocellulose (c). Adapted from [141].
Figure 7.
Structure of modified nanocellulose (CNC–UPy) by in situ polymerization (a), AFM images CNC–UPy (b), mechanical properties of native TPU and composites in the presence of modified TPU/CNC−UPy and unmodified TPU/CNC nanocellulose (c). Adapted from [141].

Figure 8.
Structure of MCNC by in situ polymerization (a), composite production (b), shape memory behaviors (i.e., shape fixity and shape recovery ratios) of TPU and composites films (c), photographs showing the thermal-induced shape memory process of the representative TPU−MCNC3 film (d), schematic showing the possible mechanism of the thermal-induced shape memory behavior of TPU−MCNC composite (e). Adapted from [130].
Figure 8.
Structure of MCNC by in situ polymerization (a), composite production (b), shape memory behaviors (i.e., shape fixity and shape recovery ratios) of TPU and composites films (c), photographs showing the thermal-induced shape memory process of the representative TPU−MCNC3 film (d), schematic showing the possible mechanism of the thermal-induced shape memory behavior of TPU−MCNC composite (e). Adapted from [130].

Figure 9.
Schematic diagram representing nanocellulose induced nucleation point to the formation of the microstructure of the hard domain in TPU. Adapted from [159]. TPU (a), TPU/nanocellulose (b–d).
Figure 9.
Schematic diagram representing nanocellulose induced nucleation point to the formation of the microstructure of the hard domain in TPU. Adapted from [159]. TPU (a), TPU/nanocellulose (b–d).

Figure 10.
Diagram showing recycling from mixed materials of nanocellulose and CNTs to applications. Adapted from [34].
Figure 10.
Diagram showing recycling from mixed materials of nanocellulose and CNTs to applications. Adapted from [34].

Figure 11.
Schematic illustration of the hydrogen bonding interaction mechanism among molecules of TPU, CNF-C, and CNTs and the film formation of TPU/CNF-C/CNTs (a), measured mechanical properties of nanocomposite films: typical tensile stress–strain curves, the inset shows the stress–strain curve of each sample stretching up to 30% (b), the strain and stress values at break for TPU/CNF-C/CNT composites with different CNT concentrations (c), experimental cyclic tensile TPU/CNF-C/CNT composites film without using any stimulus (d), and cyclic tensile of TPU/CNF-C/CNT composites film under water stimulus (e). Adapted from [25].
Figure 11.
Schematic illustration of the hydrogen bonding interaction mechanism among molecules of TPU, CNF-C, and CNTs and the film formation of TPU/CNF-C/CNTs (a), measured mechanical properties of nanocomposite films: typical tensile stress–strain curves, the inset shows the stress–strain curve of each sample stretching up to 30% (b), the strain and stress values at break for TPU/CNF-C/CNT composites with different CNT concentrations (c), experimental cyclic tensile TPU/CNF-C/CNT composites film without using any stimulus (d), and cyclic tensile of TPU/CNF-C/CNT composites film under water stimulus (e). Adapted from [25].

Figure 12.
Proposed rapidly switchable water-sensitive shape-memory mechanism for the TPU/CNCs comprising a CNC percolation network in an elastomeric matrix. Adapted from [187].
Figure 12.
Proposed rapidly switchable water-sensitive shape-memory mechanism for the TPU/CNCs comprising a CNC percolation network in an elastomeric matrix. Adapted from [187].

Figure 13.
The grasp of the plastic target object from water (a) and the release of dye (methyl blue) in water (b), the dependence of Rf on CNF content for composite films in the first (pre-stretch) and second testing cycle (c), the variation of Rr with the immersion time for different composite films in first (pre-stretch) and second testing cycle (d), the statistic of Rf and Rr of the TPU/CNF composite film containing 30% CMF for 10 times cycles, indicating that the obtained film has good recyclability (e), the schematic illustration of the test process for water-induced SMP (f). Adapted from [187].
Figure 13.
The grasp of the plastic target object from water (a) and the release of dye (methyl blue) in water (b), the dependence of Rf on CNF content for composite films in the first (pre-stretch) and second testing cycle (c), the variation of Rr with the immersion time for different composite films in first (pre-stretch) and second testing cycle (d), the statistic of Rf and Rr of the TPU/CNF composite film containing 30% CMF for 10 times cycles, indicating that the obtained film has good recyclability (e), the schematic illustration of the test process for water-induced SMP (f). Adapted from [187].

Figure 14.
Structure of the TPU/CNC nanocomposites with twin switches. Adapted from [188].
Figure 14.
Structure of the TPU/CNC nanocomposites with twin switches. Adapted from [188].

Figure 15.
Preparation of double-network TPU/NCG nanocomposites. NCG hydrogel is solvent-exchanged with acetone, THF, and then a mixture of TPU prepolymer and MOCA to form NCG-TPU prepolymer-MOCA gel. In situ polymerization yielded the TPU/NCG nanocomposite (a), schematic illustration of the synthetic procedure of the TPU/NCG nanocomposite (b), and typical stress−strain curves of neat TPU and TPU/NCG nanocomposites containing 9 vol% NCG (c). Adapted from [158].
Figure 15.
Preparation of double-network TPU/NCG nanocomposites. NCG hydrogel is solvent-exchanged with acetone, THF, and then a mixture of TPU prepolymer and MOCA to form NCG-TPU prepolymer-MOCA gel. In situ polymerization yielded the TPU/NCG nanocomposite (a), schematic illustration of the synthetic procedure of the TPU/NCG nanocomposite (b), and typical stress−strain curves of neat TPU and TPU/NCG nanocomposites containing 9 vol% NCG (c). Adapted from [158].

Figure 16.
Schematic representation of the pH-responsive shape memory materials, which rely on a hydrogen bonding switching mechanism in the interactions between cellulose nanocrystals (CNC−C6H4NO2) within polymer matrix upon immersion in HCl solution (pH = 4) or NaOH solution (pH = 8). Adapted from [23].
Figure 16.
Schematic representation of the pH-responsive shape memory materials, which rely on a hydrogen bonding switching mechanism in the interactions between cellulose nanocrystals (CNC−C6H4NO2) within polymer matrix upon immersion in HCl solution (pH = 4) or NaOH solution (pH = 8). Adapted from [23].

Figure 17.
Fabrication procedure of dual responsive smart wool fabrics functionalized by nanocomposite polymer (TPU polymer and CNC particles). Adapted from [197].
Figure 17.
Fabrication procedure of dual responsive smart wool fabrics functionalized by nanocomposite polymer (TPU polymer and CNC particles). Adapted from [197].

Figure 18.
Schematic representation of the possible flame-retardant mechanism of composite. Adapted from [130].
Figure 18.
Schematic representation of the possible flame-retardant mechanism of composite. Adapted from [130].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Standard terms for CNMs (according to TAPPI WI 3021 [55]).
Table 1.
Standard terms for CNMs (according to TAPPI WI 3021 [55]).
| Terminology and Nomenclature of Cellulose Nanomaterials | Diameter (nm) | Length (nm) | Aspect Ratio (Length/Diameter) |
|---|---|---|---|
| Cellulose NanoCrystal (CNC) | 3–10 | <500 | ˃5 |
| Cellulose NanoFibril (CNF) | 5–30 | 100–600 | ˃50 |
| Bacterial NanoCellulose (BNC) | 10–40 | ˃1000 | 100–150 |
| Cellulose MicroCrystal (CMC) | 10,000–15,000 | ˃1000 | <2 |
| Cellulose MicroFibril (CMF) | 10–100 | 500–10,000 | 50–500 |
| Amorphous NanoCellulose (ANC) | 80–120 | - | - |
| Cellulose NanoYarn (CNY) | 100–1000 | several microns | - |
| Amorphous cellulose NanoParticles (ANP) | 3 | - | - |
Table 2.
Comparison of shape recovery rates in water for TPU composites with SME containing nanocellulose.
Table 2.
Comparison of shape recovery rates in water for TPU composites with SME containing nanocellulose.
| Composite and Method of Production | Composition of Thermoplastic Polyurethanes | Nanoparticle Size and Concentration | Film Thickness (mm) | Response Temperature (°C) | Response Time (min) | Reference |
|---|---|---|---|---|---|---|
| Texin 985/CNF mixing in solution | PTMG; MDI; 1,4-butane diol | datdiameter 15 nm, length 250 nm, aspect ratio 16.67 30 wt% | 0.1–0.2 | room | 1 | [187] |
| TPU/CNC mixing in solution | PCL (Mn ~4000 Da); MDI; 1,4-butane diol | - | - | 65 | 2–3 | [188] |
| TPU HT751/CNC mixing in solution | polymethyl methacrylate | diameter 18.5 ± 5.9 nm, length 272 ± 87 nm, aspect ratio 15, 4.6 wt% | 0.2 | room | 10 | [186] |
| TPU HT-751/CNC electrospinning | polymethyl methacrylate | diameter 50–650 nm, 24.0 wt% width 200–300 nm | - | room | 2 | [185] |
| TPU-CNC In situ | HMDI; glycerol, sebacic acid | length <200 nm, aspect ratio 11.8, 18.6 vol% width 18 nm, | 0.15–0.25 | 37 (pH = 7.4), 22 (water) | 30 | [182] |
| Texin 985-CNC In situ | PTMG (Mn ~2000 Da), MDI; 1,4-butane diol | length 200 nm, aspect ratio ~11, 20 wt% | 0.2–0.3 | 37 | 24 h | [20] |
| TPU-CNF In situ | PEG (Mw ~2000 Da); TDI; 3,3-dichloro-4,4-diamino-diphenylmethanelithium | diameter 20 nm 9 wt% | - | 37 | 1 | [158] |
PTMG—poly(tetramethylene ether) glycol; MDI—4,4′-methylenediphenyl diisocyanate; PCL—poly(ε-caprolactone); HMDI—1,6-hexamethylene diisocyanate; PEG—polyethylene glycol.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gorbunova, M.; Grunin, L.; Morris, R.H.; Imamutdinova, A. Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect. J. Compos. Sci. 2023, 7, 168. https://doi.org/10.3390/jcs7040168
AMA Style
Gorbunova M, Grunin L, Morris RH, Imamutdinova A. Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect. Journal of Composites Science. 2023; 7(4):168. https://doi.org/10.3390/jcs7040168
Chicago/Turabian StyleGorbunova, Marina, Leonid Grunin, Robert H. Morris, and Arina Imamutdinova. 2023. "Nanocellulose-Based Thermoplastic Polyurethane Biocomposites with Shape Memory Effect" Journal of Composites Science 7, no. 4: 168. https://doi.org/10.3390/jcs7040168