
An Investigation of Iodovanadinite Wasteforms for the Immobilisation of Radio-Iodine and Technetium
 , , ,
, , ,
Abstract
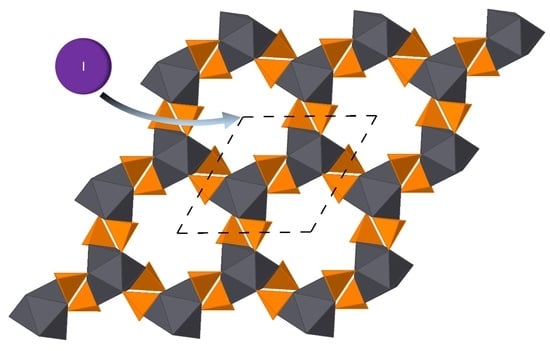
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. HIPing of Baseline Pb5(VO4)3I and the Effect of Overpressure during Consolidation
3.2. Pb5−xAgx(VO4)3−x(MoO4)xI Series
3.3. Pb4.5−xBaxAg0.5(VO4)3I0.5 Series
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wilson, P.D. The Nuclear Fuel Cycle: From Ore to Waste; Oxford University Press: Oxford, UK, 1996. [Google Scholar]
- Riley, B.J.; Vienna, J.D.; Strachan, D.M.; McCloy, J.S.; Jerden, J.L., Jr. Materials and processes for the effective capture and immobilization of radioiodine: A review. J. Nucl. Mater. 2016, 470, 307–326. [Google Scholar] [CrossRef]
- O’Sullivan, S.E.; Montoya, E.; Sun, S.K.; George, J.; Kirk, C.; Dixon Wilkins, M.C.; Weck, P.F.; Kim, E.; Knight, K.S.; Hyatt, N.C. Crystal and electronic structures of A2NaIO6 periodate double perovskites (A = Sr, Ca, Ba): Candidate wasteforms for I-129 immobilization. Inorg. Chem. 2020, 59, 18407–18419. [Google Scholar] [CrossRef]
- Asmussen, R.M.; Turner, J.; Chong, S.; Riley, B.J. Review of recent developments in iodine wasteform production. Front. Chem. 2022, 10, 1043653. [Google Scholar] [CrossRef] [PubMed]
- Umadevi, K.; Mandal, D. Performance of radio-iodine discharge control methods of nuclear reprocessing plants. J. Environ. Radioact. 2021, 234, 106623. [Google Scholar] [CrossRef] [PubMed]
- Buck, E.C.; Mausolf, E.J.; McNamara, B.K.; Soderquist, C.Z.; Schwantes, J.M. Sequestration of radioactive iodine in silver-palladium phases in commercial spent nuclear fuel. J. Nucl. Mater. 2016, 482, 229–235. [Google Scholar] [CrossRef]
- Chapman, K.W.; Chupas, P.J.; Nenoff, T.M. Radioactive iodine capture in silver-containing mordenites through nanoscale silver iodide formation. J. Am. Chem. Soc. 2010, 132, 8897–8899. [Google Scholar] [CrossRef]
- Olsen, Y.S.; i Batlle, J.V. A model for the bioaccumulation of 99Tc in lobsters (Homarus gammarus) from the West Cumbrian coast. J. Environ. Radioact. 2003, 67, 219–233. [Google Scholar] [CrossRef]
- Donald, I.W. Waste Immobilization in Glass and Ceramic Based Hosts: Radioactive, Toxic and Hazardous Wastes; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Schwochau, K. Technetium: Chemistry and Radiopharmaceutical Applications; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Poineau, F.; Du Mazaubrun, J.; Ford, D.; Fortner, J.; Kropf, J.; Silva, G.C.; Smith, N.; Long, K.; Jarvinen, G.; Czerwinski, K.R. Uranium/technetium separation for the UREX process–synthesis and characterization of solid reprocessing forms. Radiochim. Acta 2008, 96, 527–533. [Google Scholar] [CrossRef]
- Audubert, F.; Carpena, J.; Lacout, J.L.; Tetard, F. Elaboration of an iodine-bearing apatite iodine diffusion into a Pb3(VO4)2 matrix. Solid State Ionics 1997, 95, 113–119. [Google Scholar] [CrossRef]
- White, T.J.; d Dong, Z. Structural derivation and crystal chemistry of apatites. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2003, 59, 1–16. [Google Scholar] [CrossRef]
- Schriewer, M.S.; Jeitschko, W. Preparation and crystal structure of the isotypic orthorhombic strontium perrhenate halides Sr5(ReO5)3X (X = Cl, Br, I) and structure refinement of the related hexagonal apatite-like compound Ba5(ReO5)3Cl. J. Solid State Chem. 1993, 107, 1–11. [Google Scholar] [CrossRef]
- Audubert, F.; Savariault, J.M.; Lacout, J.L. Pentalead tris (vanadate) iodide, a defect vanadinite-type compound. Acta Crystallogr. C Struct. Chem. 1999, 55, 271–273. [Google Scholar] [CrossRef]
- Baud, G.; Besse, J.P.; Sueur, G.; Chevalier, R. Structure de nouvelles apatites au rhenium contenant des anions volumineux: Ba10(ReO5)6X2 (X = Br, I). Mater. Res. Bull. 1979, 14, 675–682. [Google Scholar] [CrossRef]
- Le Gallet, S.; Campayo, L.; Courtois, E.; Hoffmann, S.; Grin, Y.; Bernard, F.; Bart, F. Spark plasma sintering of iodine-bearing apatite. J. Nucl. Mater. 2010, 400, 251–256. [Google Scholar] [CrossRef]
- Stennett, M.C.; Pinnock, I.J.; Hyatt, N.C. Rapid synthesis of Pb5(VO4)3I, for the immobilisation of iodine radioisotopes, by microwave dielectric heating. J. Nucl. Mater. 2011, 414, 352–359. [Google Scholar] [CrossRef]
- Redfern, S.A.T.; Smith, S.E.; Maddrell, E.R. High-temperature breakdown of the synthetic iodine analogue of vanadinite, Pb5 (VO4)3I: An apatite-related compound for iodine radioisotope immobilization? Miner. Mag. 2012, 76, 997–1003. [Google Scholar] [CrossRef]
- Suetsugu, Y. Synthesis of lead vanadate iodoapatite utilizing dry mechanochemical process. J. Nucl. Mater. 2014, 454, 223–229. [Google Scholar] [CrossRef]
- Lu, F.; Yao, T.; Xu, J.; Wang, J.; Scott, S.; Dong, Z.; Ewing, R.C.; Lian, J. Facile low temperature solid state synthesis of iodoapatite by high-energy ball milling. RSC Adv. 2014, 4, 38718–38725. [Google Scholar] [CrossRef]
- Zhang, M.; Maddrell, E.R.; Abraitis, P.K.; Salje, E.K.H. Impact of leach on lead vanado-iodoapatite [Pb5(VO4)3I]: An infrared and Raman spectroscopic study. Mater. Sci. Eng. B Solid State Mater. Adv. Technol. 2007, 137, 149–155. [Google Scholar] [CrossRef]
- Maddrell, E.R.; Abraitis, P.K. A comparison of wasteforms and processes for the immobilisation of iodine-129. MRS Proc. 2003, 807, 565–570. [Google Scholar] [CrossRef]
- Guy, C.; Audubert, F.; Lartigue, J.E.; Latrille, C.; Advocat, T.; Fillet, C. New conditionings for separated long-lived radionuclides. Comptes Rendus Phys. 2002, 3, 827–837. [Google Scholar] [CrossRef]
- Uno, M.; Kosuga, A.; Masuo, S.; Imamura, M.; Yamanaka, S. Thermal and mechanical properties of AgPb9(VO4)6I and AgBa9(VO4)6I. J. Alloys Compd. 2004, 384, 300–302. [Google Scholar] [CrossRef]
- Johnstone, E.V.; Bailey, D.J.; Stennett, M.C.; Heo, J.; Hyatt, N.C. On the existence of AgM9(VO4)6I (M = Ba, Pb). RSC Adv. 2017, 7, 49004–49009. [Google Scholar] [CrossRef]
- Yao, T.; Lu, F.; Sun, H.; Wang, J.; Ewing, R.C.; Lian, J. Bulk iodoapatite ceramic densified by spark plasma sintering with exceptional thermal stability. J. Am. Ceram. Soc. 2014, 97, 2409–2412. [Google Scholar] [CrossRef]
- Klement, R.; Harth, R. Über Bleiapatite und tertiäres Bleiphosphat. Z. Für Anorg. Allg. Chem. 1962, 314, 238–244. [Google Scholar] [CrossRef]
- Porter, S.K.; Scheckel, K.G.; Impellitteri, C.A.; Ryan, J.A. Toxic metals in the environment: Thermodynamic considerations for possible immobilization strategies for Pb, Cd, As, and Hg. Crit. Rev. Environ. Sci. Technol. 2004, 34, 495–604. [Google Scholar] [CrossRef]
- Ichikawa, F. The isolation of technetium by coprecipitation or anion exchange. Bull. Chem. Soc. Jpn. 1959, 32, 1126–1129. [Google Scholar] [CrossRef]
- Klement, R.; Harth, R. Das Verhalten von tertiären Erdalkaliphosphaten, -arsenaten und -vanadaten in geschmolzenen Halogeniden. Chem. Ber. 1961, 94, 1452–1456. [Google Scholar] [CrossRef]
- Johnstone, E.V.; Bailey, D.J.; Lawson, S.; Stennett, M.C.; Corkhill, C.L.; Kim, M.; Heo, J.; Matsumura, D.; Hyatt, N.C. Synthesis and characterization of iodovanadinite using PdI2, an iodine source for the immobilisation of radioiodine. RSC Adv. 2020, 10, 25116–25124. [Google Scholar] [CrossRef]
- Hyatt, N.C.; Corkhill, C.L.; Stennett, M.C.; Hand, R.J.; Gardner, L.J.; Thorpe, C.L. The HADES facility for high activity decommissioning engineering & science: Part of the UK national nuclear user facility. IOP Conf. Ser. Mater. Sci. 2020, 818, 012022. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Composition | a (Å) | c (Å) |
|---|---|---|
| Pb5(VO4)3I | 10.4530 (1) | 7.4822 (1) |
| Pb4.8Ag0.2(VO4)2.8(MoO4)0.2I | 10.4695 (2) | 7.4816 (2) |
| Pb4.6Ag0.4(VO4)2.6(MoO4)0.4I | 10.464 (1) | 7.483 (1) |
| Pb4.4Ag0.6(VO4)2.4(MoO4)0.6I | - | - |
| Pb4.2Ag0.8(VO4)2.2(MoO4)0.8I | - | - |
| Pb4.0Ag1.0(VO4)2.0(MoO4)1.0I | - | - |
| Target Composition | Ba3(VO4)2 Phase EDX Composition | a (Å) | α (º) |
|---|---|---|---|
| Pb4.5Ag0.5(VO4)3I0.5 | Pb2.96±0.15Ag0.04±0.06(VO4)2.24±0.17I0.06±0.05 | - | - |
| Pb4.0Ba0.5Ag0.5(VO4)3I0.5 | Pb2.55±0.09Ba0.44±0.04Ag0.01±0.02(VO4)2.08±0.06I0.03±0.04 | 7.590 (2) | 44.756 (8) |
| Pb3.5Ba0.5Ag0.5(VO4)3I0.5 | Pb2.16±0.13Ba0.75±0.33Ag0.1±0.05(VO4)1.39±0.32I0.03±0.03 | 7.642 (1) | 44.525 (7) |
| Pb3.0Ba1.5Ag0.5(VO4)3I0.5 | Pb1.94±0.13Ba0.99±0.05Ag0.07±0.06(VO4)1.81±0.18I0.01±0.02 | 7.689 (1) | 44.259 (5) |
| Pb2.5Ba2.0Ag0.5(VO4)3I0.5 | Pb1.69±0.27Ba1.24±0.14Ag0.07±0.06(VO4)1.92±0.24I0.03±0.04 | 7.733 (1) | 44.010 (6) |
| Pb2.0Ba2.5Ag0.5(VO4)3I0.5 | Pb1.30±0.08Ba1.63±0.09Ag0.07±0.05(VO4)1.78±0.07I0.01±0.02 | 7.769 (1) | 43.771 (5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailey, D.J.; Johnstone, E.V.; Stennett, M.C.; Corkhill, C.L.; Hyatt, N.C. An Investigation of Iodovanadinite Wasteforms for the Immobilisation of Radio-Iodine and Technetium. Ceramics 2023, 6, 1826-1839. https://doi.org/10.3390/ceramics6030111
Bailey DJ, Johnstone EV, Stennett MC, Corkhill CL, Hyatt NC. An Investigation of Iodovanadinite Wasteforms for the Immobilisation of Radio-Iodine and Technetium. Ceramics. 2023; 6(3):1826-1839. https://doi.org/10.3390/ceramics6030111
Chicago/Turabian StyleBailey, Daniel J., Erik V. Johnstone, Martin C. Stennett, Claire L. Corkhill, and Neil C. Hyatt. 2023. "An Investigation of Iodovanadinite Wasteforms for the Immobilisation of Radio-Iodine and Technetium" Ceramics 6, no. 3: 1826-1839. https://doi.org/10.3390/ceramics6030111