Nuclear Magnetic Resonance Analysis of Changes in Dissolved Organic Matter Composition with Successive Layering on Clay Mineral Surfaces


Abstract
:1. Introduction
2. Materials and Methods
2.1. Clay Mineral and DOM Samples
2.2. Preparation of Organo-Mineral Complexes
2.3. NMR Spectroscopy Experiments
3. Results
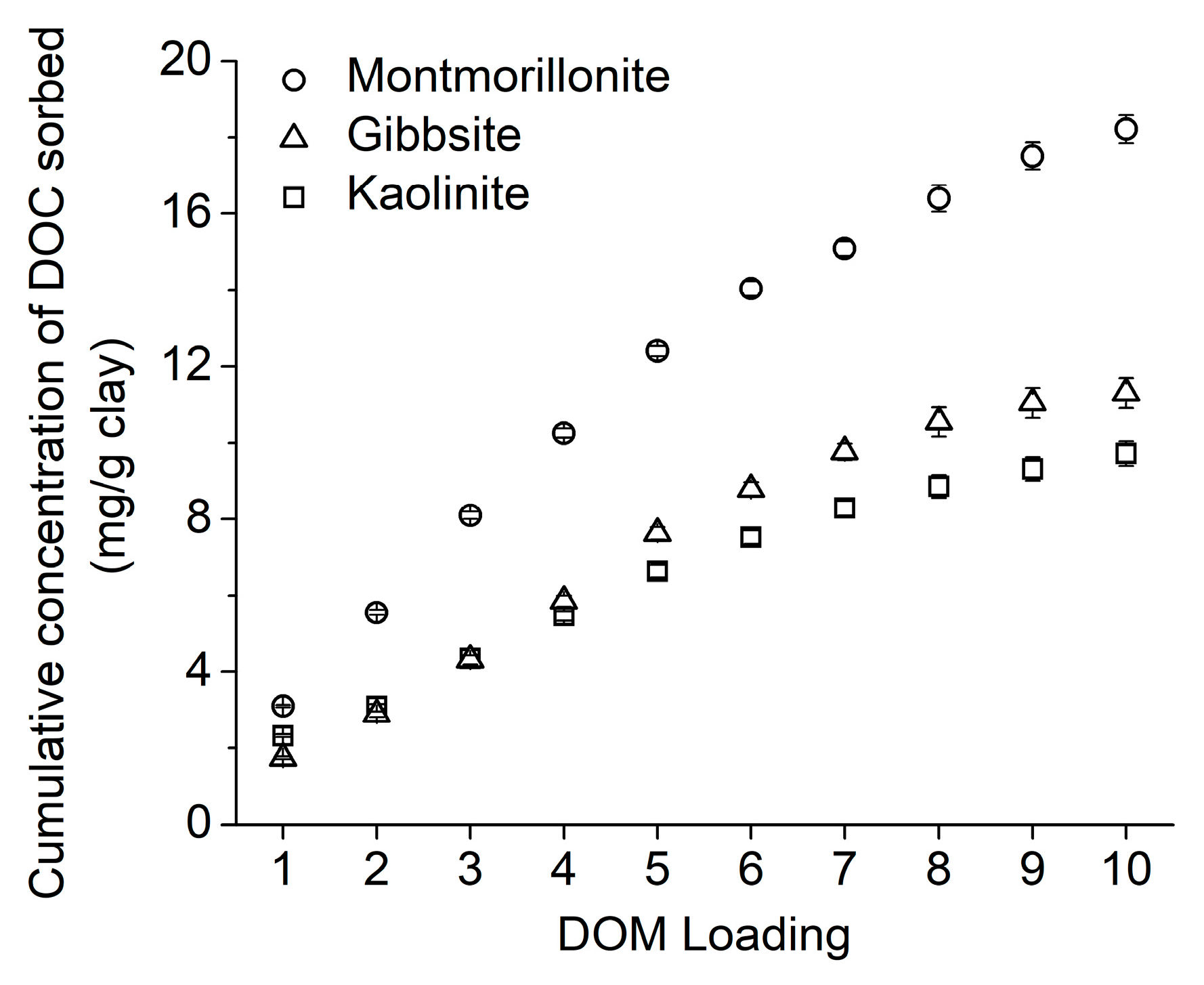
3.1. Dissolved Organic Matter Sorption to Clay Minerals
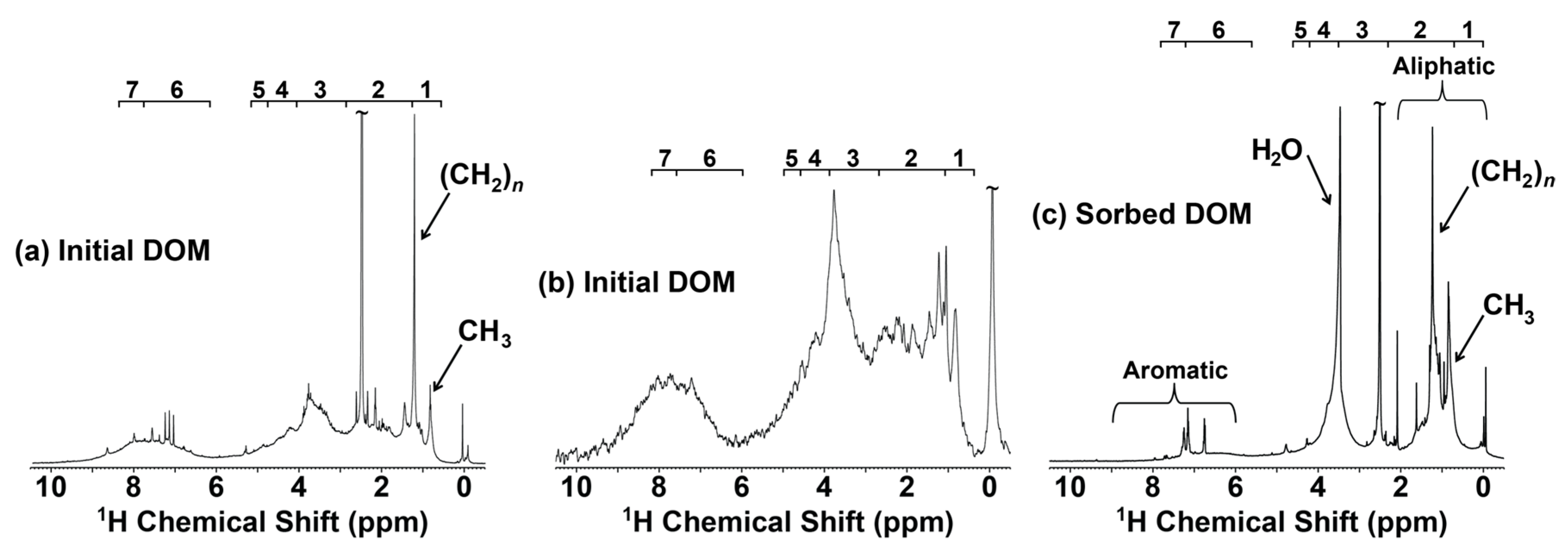
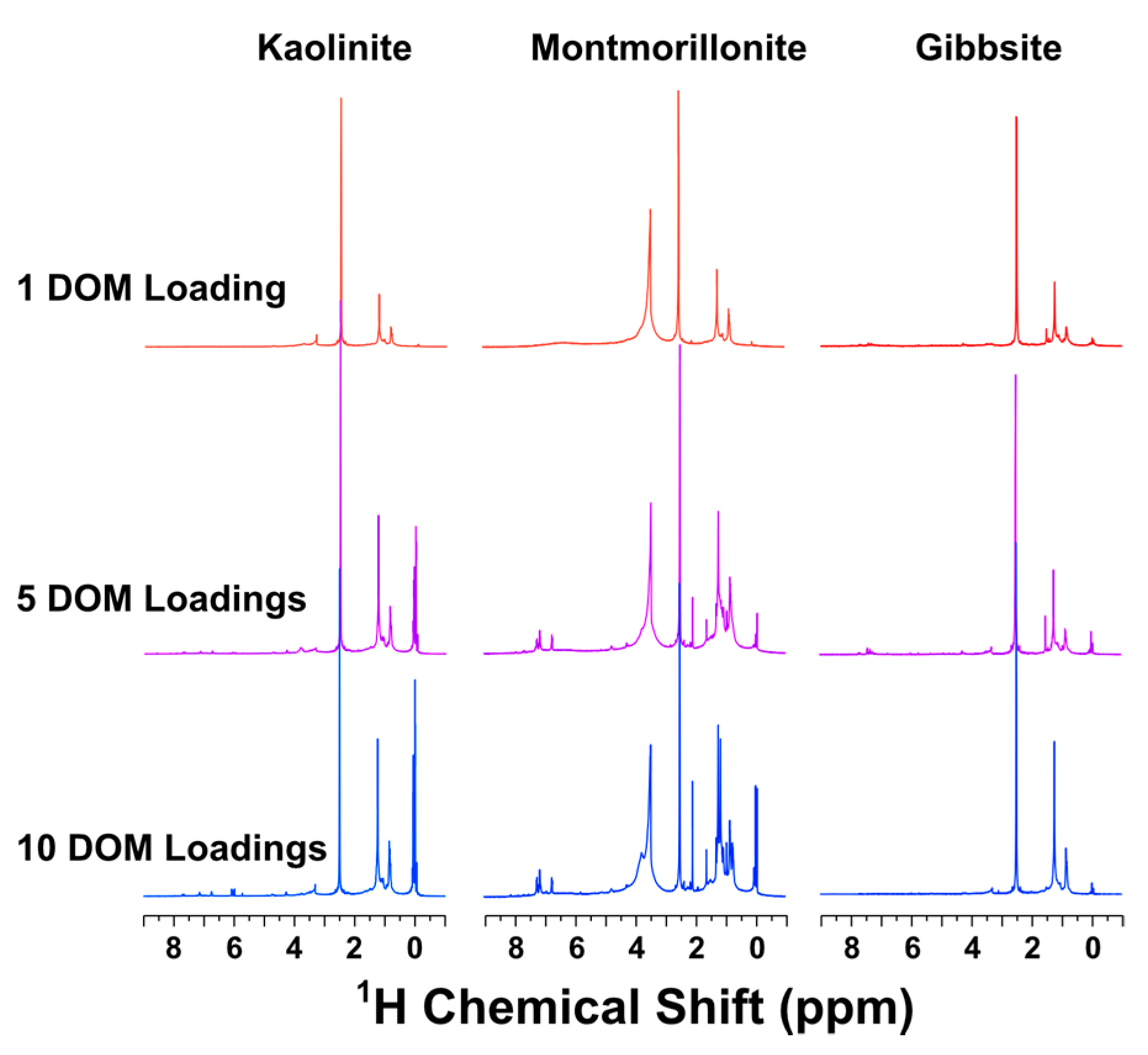
3.2. NMR Characterization of Unsorbed DOM and Organo-Clay Complexes
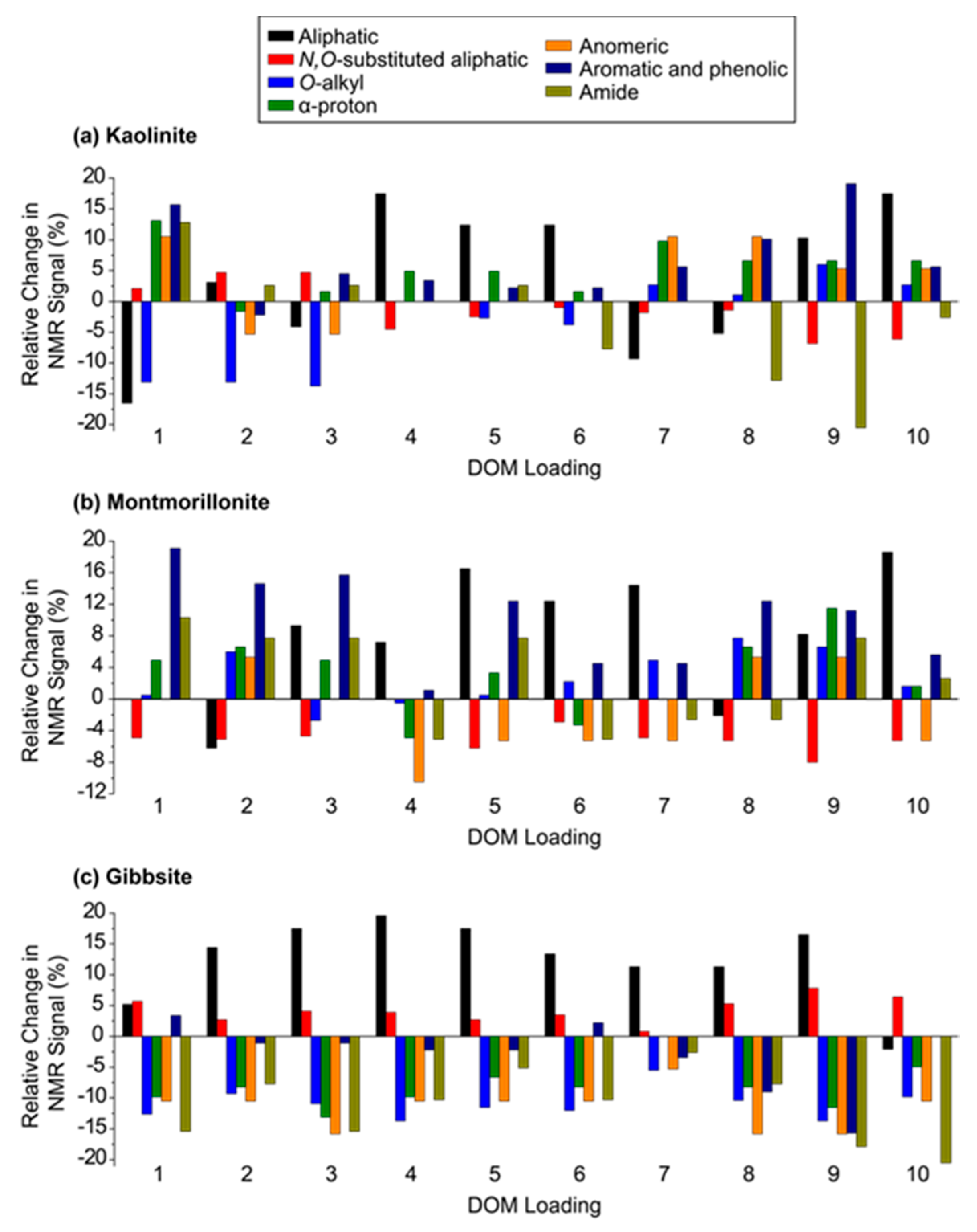
3.3. Sorption of Specific DOM Components to Kaolinite
3.4. Sorption of Specific DOM Components to Montmorillonite
3.5. Sorption of Specific DOM Components to Gibbsite
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lal, R. Sequestration of atmospheric CO2 in global carbon pools. Energy Environ. Sci. 2008, 1, 86–100. [Google Scholar] [CrossRef]
- Schmidt, M.W.I.; Torn, M.S.; Abiven, S.; Dittmar, T.; Guggenberger, G.; Janssens, I.A.; Kleber, M.; Kögel-Knabner, I.; Lehmann, J.; Manning, D.A.C.; et al. Persistence of soil organic matter as an ecosystem property. Nature 2011, 478, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.; Sollins, P.; Sutton, R. A conceptual model of organo-mineral interactions in soils: Self-assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry 2007, 85, 9–24. [Google Scholar] [CrossRef]
- Lin, L.H.; Simpson, M.J. Enhanced extractability of cutin- and suberin-derived organic matter with demineralization implies physical protection over chemical recalcitrance in soil. Org. Geochem. 2016, 97, 111–121. [Google Scholar] [CrossRef]
- Torn, M.S.; Trumbore, S.E.; Chadwick, O.A.; Vitousek, P.M.; Hendricks, D.M. Mineral control of soil organic carbon storage and turnover. Nature 1997, 389, 170–173. [Google Scholar] [CrossRef]
- Von Lützow, M.; Kögel-Knabner, I.; Ekschmitt, K.; Matzner, E.; Guggenberger, G.; Marschner, B.; Flessa, H. Stabilization of organic matter in temperate soils: Mechanisms and their relevance under different soil conditions—A review. Eur. J. Soil Sci. 2006, 57, 426–445. [Google Scholar] [CrossRef]
- Ghosh, S.; Wang, Z.; Kang, S.; Bhowmik, P.C.; Xing, B. Sorption and fractionation of a peat derived humic acid by kaolinite, montmorillonite, and goethite. Pedosphere 2009, 19, 21–30. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G.; Haumaier, L.; Zech, W. Dissolved organic matter sorption on subsoils and minerals studied by 13C NMR and DRIFT spectroscopy. Eur. J. Soil Sci. 1997, 48, 301–310. [Google Scholar] [CrossRef]
- Meier, M.; Namjesnik-Dejanovic, K.; Maurice, P.A.; Chin, Y.; Aiken, G.R. Fractionation of aquatic natural organic matter upon sorption to goethite and kaolinite. Chem. Geol. 1999, 157, 275–284. [Google Scholar] [CrossRef]
- Mitchell, P.J.; Simpson, A.J.; Soong, R.; Oren, A.; Chefetz, B.; Simpson, M.J. Solution-state NMR investigation of the sorptive fractionation of dissolved organic matter by alkaline mineral soils. Environ. Chem. 2013, 10, 333–340. [Google Scholar] [CrossRef]
- Sanderman, J.; Maddern, T.; Baldock, J. Similar composition but differential stability of mineral retained organic matter across four classes of clay minerals. Biogeochemistry 2014, 121, 409–424. [Google Scholar] [CrossRef]
- Gu, B.; Schmitt, J.; Chen, Z.; Liang, L.; McCarthy, J.F. Adsorption and desorption of natural organic matter on iron oxide-mechanisms and models. Environ. Sci. Technol. 1994, 28, 38–46. [Google Scholar] [CrossRef] [PubMed]
- McKnight, D.M.; Bencala, K.E.; Zellweger, G.W.; Aiken, G.R.; Feder, G.L.; Thorn, K.A. Sorption of dissolved organic carbon by hydrous aluminum and iron oxides occurring at the confluence of Deer Creek with the Snake River, Summit County, Colorado. Environ. Sci. Technol. 1992, 26, 1388–1396. [Google Scholar] [CrossRef]
- Feng, X.; Simpson, A.J.; Simpson, M.J. Chemical and mineralogical controls on humic acid sorption to clay mineral surfaces. Org. Geochem. 2005, 36, 1553–1566. [Google Scholar] [CrossRef]
- Genest, S.C.; Simpson, M.J.; Simpson, A.J.; Soong, R.; McNally, D.J. Analysis of soil organic matter at the solid-water interface by nuclear magnetic resonance spectroscopy. Environ. Chem. 2014, 11, 472–482. [Google Scholar] [CrossRef]
- Wang, K.; Xing, B. Structural and sorption characteristics of adsorbed humic acid on clay minerals. J. Environ. Qual. 2005, 34, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Sollins, P.; Kramer, M.G.; Swanston, C.; Lajtha, K.; Filley, T.; Aufdenkampe, A.K.; Wagai, R.; Bowden, R.D. Sequential density fractionation across soils of contrasting mineralogy: Evidence for both microbial- and mineral-controlled soil organic matter stabilization. Biogeochemistry 2009, 96, 209–231. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G. Mineral surfaces and soil organic matter. Eur. J. Soil Sci. 2003, 54, 219–236. [Google Scholar] [CrossRef]
- De Junet, A.; Basile-Doelsch, I.; Borschneck, D.; Masion, A.; Legros, S.; Marol, C.; Balesdent, J.; Templier, J.; Derenne, S. Characterisation of organic matter from organo-mineral complexes in an Andosol from Reunion island. J. Anal. Appl. Pyrol. 2013, 99, 92–100. [Google Scholar] [CrossRef]
- Kaiser, K.; Guggenberger, G.; Zech, W. Sorption of DOM and DOM fractions to forest soils. Geoderma 1996, 74, 281–303. [Google Scholar] [CrossRef]
- Mayer, L.M. Relationships between mineral surfaces and organic carbon concentrations in soils and sediments. Chem. Geol. 1994, 114, 347–363. [Google Scholar] [CrossRef]
- Nelson, P.N.; Baldock, J.A.; Oades, J.M. Concentration and composition of dissolved organic carbon in streams in relation to catchment soil properties. Biogeochemistry 1992, 19, 27–50. [Google Scholar] [CrossRef]
- Mikutta, R.; Kleber, M.; Torn, M.S.; Jahn, R. Stabilization of soil organic matter: Association with minerals or chemical recalcitrance. Biogeochemistry 2006, 77, 25–56. [Google Scholar] [CrossRef]
- Clemente, J.S.; Simpson, M.J. Physical protection of lignin by organic matter and clay minerals from chemical oxidation. Org. Geochem. 2013, 58, 1–12. [Google Scholar] [CrossRef]
- Thevenot, M.; Dignac, M.; Mendez-Millan, M.; Bahri, H.; Hatté, C.; Bardoux, G.; Rumpel, C. Ligno-aliphatic complexes in soils revealed by an isolation procedure: Implication for lignin fate. Biol. Fert. Soils 2013, 49, 517–526. [Google Scholar] [CrossRef]
- Zang, X.; van Heemst, J.D.H.; Dria, K.J.; Hatcher, P.G. Encapsulation of protein in humic acid from a Histosol as an explanation for the occurrence of organic nitrogen in soil and sediment. Org. Geochem. 2000, 31, 679–695. [Google Scholar] [CrossRef]
- Mitchell, P.J.; Simpson, M.J. High affinity sorption domains in soil are blocked by polar soil organic matter components. Environ. Sci. Technol. 2013, 47, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Masoom, H.; Courtier-Murias, D.; Farooq, H.; Soong, R.; Kelleher, B.P.; Zhang, C.; Maas, W.E.; Fey, M.; Kumar, R.; Monette, M.; et al. Soil organic matter in its native state: Unravelling the most complex biomaterial on earth. Environ. Sci. Technol. 2016, 50, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.J.; Kingery, W.L.; Shaw, D.R.; Spraul, M.; Humpfer, E.; Dvortsak, P. The application of 1H HR-MAS NMR spectroscopy for the study of structures and associations of organic components at the solid-aqueous interface of a whole soil. Environ. Sci. Technol. 2001, 35, 3321–3325. [Google Scholar] [CrossRef] [PubMed]
- Arnarson, T.S.; Keil, R.G. Mechanisms of pore water organic matter adsorption to montmorillonite. Mar. Chem. 2000, 71, 309–320. [Google Scholar] [CrossRef]
- Tombácz, E.; Libor, Z.; Illés, E.; Majzik, A.; Klumpp, E. The role of reactive surface sites and complexation by humic acids in the interaction of clay mineral and iron oxide particles. Org. Geochem. 2004, 35, 257–267. [Google Scholar] [CrossRef]
- Dufrêne, Y.F.; Boonaert, C.J.; Rouxhet, P.G. Adhesion of azospirillum brasilense: Role of proteins at the cell-support interface. Colloids Surf. B 1996, 7, 113–128. [Google Scholar] [CrossRef]
- Wershaw, R.L.; Llaguno, E.C.; Leenheer, J.A. Mechanism of formation of humus coatings on mineral surfaces 3. Composition of adsorbed organic acids from compost leachate on alumina by solid-state 13C nmr. Colloids Surf. A 1996, 108, 213–223. [Google Scholar] [CrossRef]
- Almendros, G.; Guadalix, M.E.; González-Vila, F.J.; Martin, F. Preservation of aliphatic macromolecules in soil humins. Org. Geochem. 1996, 24, 651–659. [Google Scholar] [CrossRef]
- Sollins, P.; Homann, P.; Caldwell, B.A. Stabilization and destabilization of soil organic matter: Mechanisms and controls. Geoderma 1996, 74, 65–105. [Google Scholar] [CrossRef]
- Van Olphen, H.; Fripiat, J.J. Data Handbook for Clay Minerals and Other Non-Metallic Materials; Pergamon Press: Oxford, UK, 1979; p. 346. [Google Scholar]
- Farooq, H.; Courtier-Murias, D.; Soong, R.; Bermel, W.; Kingery, W.M.; Simpson, A.J. HR-MAS NMR spectroscopy: A practical guide for natural samples. Curr. Org. Chem. 2014, 17, 3013–3031. [Google Scholar] [CrossRef]
- Rhoades, J.D. Cation Exchange Capacity. Methods of Soil Analysis, 2nd ed.; Soil Science Society of America: Madison, WI, USA, 1982; pp. 149–157. [Google Scholar]
- Salloum, M.J.; Dudas, M.J.; McGill, W.B. Variation of 1-naphthol sorption with organic matter fractionation: The role of physical conformation. Org. Geochem. 2001, 32, 709–719. [Google Scholar] [CrossRef]
- Simpson, A.J.; Song, G.; Smith, E.; Lam, B.; Novotny, E.H.; Hayes, M.H.B. Unraveling the structural components of soil humin by use of solution-state nuclear magnetic resonance spectroscopy. Environ. Sci. Technol. 2007, 41, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.H.; Chen, A.D.; Johnson, C.S. An improved diffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Kelleher, B.P.; Simpson, A.J. Humic substances in soils: Are they really chemically distinct? Environ. Sci. Technol. 2006, 40, 4605–4611. [Google Scholar] [CrossRef] [PubMed]
- Pautler, B.G.; Dubnick, A.; Sharp, M.J.; Simpson, A.J.; Simpson, M.J. Comparison of cryoconite organic matter composition from Arctic and Antarctic glaciers at the molecular-level. Geochim. Cosmochim. Acta 2013, 104, 1–18. [Google Scholar] [CrossRef]
- Pisani, O.; Frey, S.D.; Simpson, A.J.; Simpson, M.J. Soil warming and nitrogen deposition alter soil organic matter composition at the molecular-level. Biogeochemistry 2015, 123, 391–409. [Google Scholar] [CrossRef]
- Clemente, J.S.; Gregorich, E.G.; Simpson, A.J.; Kumar, R.; Courtier-Murias, D.; Simpson, M.J. Comparison of nuclear magnetic resonance methods for the analysis of organic matter composition from soil density and particle fractions. Environ. Chem. 2012, 9, 97–107. [Google Scholar] [CrossRef]
- Courtier-Murias, D.; Farooq, H.; Masoom, H.; Botana, A.; Soong, R.; Longstaffe, J.G.; Simpson, M.J.; Maas, W.E.; Fey, M.; Andrew, B.; et al. Comprehensive multiphase NMR spectroscopy: Basic experimental approaches to differentiate phases in heterogeneous samples. J. Magn. Reson. 2012, 217, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Farooq, H.; Courtier-Murias, D.; Simspon, M.J.; Maas, W.E.; Fey, M.; Andrew, B.; Struppe, J.; Hutchins, H.; Krishnamurthy, S.; Kumar, R.; et al. Characterisation of oil contaminated soils by comprehensive multiphase NMR spectroscopy. Environ. Chem. 2015, 12, 227–235. [Google Scholar] [CrossRef]
- Kahle, M.; Kleber, M.; Jahn, R. Carbon storage in loess derived surface soils from central Germany: Influence of mineral phase variables. J. Plant Nutr. Soil Sci. 2002, 165, 141–149. [Google Scholar] [CrossRef]
- Duarte-Silva, R.; Villa-García, M.A.; Rendueles, M.; Díaz, M. Structural, textural and protein adsorption properties of kaolinite and surface modified kaolinite adsorbents. Appl. Clay Sci. 2014, 90, 73–80. [Google Scholar] [CrossRef]
- Fiorito, T.M.; Icoz, I.; Stotzky, G. Adsorption and binding of the transgenic plant proteins, human serum albumin, β-glucuronidase, and cry3bb1, on montmorillonite and kaolinite: Microbial utilization and enzymatic activity of free and clay-bound proteins. Appl. Clay Sci. 2008, 39, 142–150. [Google Scholar] [CrossRef]
- Hlady, V.; Buijs, J. Protein adsorption on solid surfaces. Curr. Opin. Biotechnol. 1996, 7, 72–77. [Google Scholar] [CrossRef]
- Stotzky, G. Influence of soil mineral colloids on metabolic processes, growth, adhesion, and ecology of microbes and viruses. In Interactions of Soil Minerals with Natural Organics and Microbes; Huang, P., Schnitzer, M., Eds.; Soil Science Society of America: Madison, WI, USA, 1986; Volume 17, pp. 305–428. [Google Scholar]
- Kögel-Knabner, I.; Amelung, W. Dynamics, chemistry, and preservation of organic matter in soils. Treatise Geochem. 2014, 12, 157–215. [Google Scholar]
- Tonneijck, F.H.; Jansen, B.; Nierop, K.G.J.; Verstraten, J.M.; Sevink, J.; De Lange, L. Towards understanding of carbon stocks and stabilization in volcanic ash soils in natural Andean ecosystems of northern Ecuador. Eur. J. Soil Sci. 2010, 61, 392–405. [Google Scholar] [CrossRef]
- Schöning, I.; Knicker, H.; Kögel-Knabner, I. Intimate association between O/N-alkyl carbon and iron oxides in clay fractions of forest soils. Org. Geochem. 2005, 36, 1378–1390. [Google Scholar] [CrossRef]
- Oren, A.; Chefetz, B. Sorptive and desorptive fractionation of dissolved organic matter by mineral soil matrices. J. Environ. Qual. 2012, 41, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, A. The supramolecular structure of humic substances. Soil Sci. 2001, 166, 810–832. [Google Scholar] [CrossRef]
- Schulten, H.; Schnitzer, M. Chemical model structures for soil organic matter and soils. Soil Sci. 1997, 162, 115–130. [Google Scholar] [CrossRef]
- Baldock, J.A.; Skjemstad, J.O. Role of the soil matrix and minerals in protecting natural organic materials against biological attack. Org. Geochem. 2000, 31, 697–710. [Google Scholar] [CrossRef]
- Kleber, M. What is recalcitrant soil organic matter? Environ. Chem. 2010, 7, 320–332. [Google Scholar] [CrossRef]
- Gleixner, G.; Poirier, N.; Bol, R.; Balesdent, J. Molecular dynamics of organic matter in a cultivated soil. Org. Geochem. 2002, 33, 357–366. [Google Scholar] [CrossRef]
- Murphy, E.M.; Zachara, J.M.; Smith, S.C.; Phillips, J.L.; Wietsma, T.W. Interaction of hydrophobic organic compounds with mineral-bound humic substances. Environ. Sci. Technol. 1994, 28, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Varadachari, C.; Mondal, A.H.; Dulal, C.N.; Ghosh, K. Clay-humus complexation: Effect of pH and the nature of bonding. Soil Biol. Biochem. 1994, 26, 1145–1149. [Google Scholar] [CrossRef]
- Chorover, J.; Amistadi, M.K. Reaction of forest floor organic matter at goethite, birnessite and smectite surfaces. Geochim. Cosmochim. Acta 2001, 65, 95–109. [Google Scholar] [CrossRef]
- Parfitt, R.L.; Childs, C.W. Estimation of forms of Fe and Al: A review, and analysis of contrasting soils by dissolution and Mössbauer methods. Aust. J. Soil Res. 1988, 26, 121–144. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Specific Surface Area (m2/g) | Cation Exchange Capacity (cmolc/kg) |
|---|---|---|
| Kaolinite 1 | 10.05 | 2.0 |
| Montmorillonite 1 | 83.79 | 84.4 |
| Gibbsite 2 | 1.41 | 0.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitchell, P.J.; Simpson, A.J.; Soong, R.; Simpson, M.J. Nuclear Magnetic Resonance Analysis of Changes in Dissolved Organic Matter Composition with Successive Layering on Clay Mineral Surfaces. Soil Syst. 2018, 2, 8. https://doi.org/10.3390/soils2010008
Mitchell PJ, Simpson AJ, Soong R, Simpson MJ. Nuclear Magnetic Resonance Analysis of Changes in Dissolved Organic Matter Composition with Successive Layering on Clay Mineral Surfaces. Soil Systems. 2018; 2(1):8. https://doi.org/10.3390/soils2010008
Chicago/Turabian StyleMitchell, Perry J., André J. Simpson, Ronald Soong, and Myrna J. Simpson. 2018. "Nuclear Magnetic Resonance Analysis of Changes in Dissolved Organic Matter Composition with Successive Layering on Clay Mineral Surfaces" Soil Systems 2, no. 1: 8. https://doi.org/10.3390/soils2010008