Optimization of Data Processing Minimizes Impact of Self-Absorption on Phosphorus Speciation Results by P K-Edge XANES

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ternary Standard Mixtures and Soil Sample
2.2. Phosphorus K-Edge XANES Data Collection and Analysis
3. Results
3.1. Analysis of P Content of the Standard Mixtures and Soil Sample
3.2. Self-Absorption Effects on LC Fitting of Standard Mixtures and Soils
3.3. Soil Sample
4. Discussion
4.1. Analysis of P Content of the Standard Mixtures and Soil Sample
4.2. Implications and Caveats for P Speciation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stern, E.A. Structure determination by X-ray absorption. Contemp. Phys. 1978, 19, 289–310. [Google Scholar] [CrossRef]
- Rehr, J.J.; Ankudinov, A.L. Progress in the theory and interpretation of XANES. Coord. Chem. Rev. 2005, 249, 131–140. [Google Scholar] [CrossRef]
- Hesterberg, D.; Zhou, W.; Hutchison, K.J.; Beauchemin, S.; Sayers, D.E. XAFS study of adsorbed and mineral forms of phosphate. J. Synchrotron Radiat. 1999, 6, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.; Leinweber, P. Phosphorus in sequentially extracted fen peat soils: A K-edge X-ray absorption near-edge structure (XANES) spectroscopy study. J. Plant Nutr. Soil Sci. 2008, 171, 613–620. [Google Scholar] [CrossRef]
- Eveborn, D.; Gustafsson, J.P.; Hesterberg, D.; Hillier, S. XANES speciation of P in environmental samples: An assessment of filter media for on-site wastewater treatment. Environ. Sci. Technol. 2009, 43, 6515–6521. [Google Scholar] [CrossRef]
- Prietzel, J.; Klysubun, W.; Werner, F. Speciation of phosphorus in temperate zone forest soils as assessed by combined wet-chemical fractionation and XANES spectroscopy. J. Plant Nutr. Soil Sci. 2016, 179, 168–185. [Google Scholar] [CrossRef]
- Abdala, D.B.; Northrup, P.A.; Vicentin, F.C.; Sparks, D.L. Residence time and pH effects on the bonding configuration of orthophosphate surface complexes at the goethite/water interface as examined by Extended X-ray Absorption Fine Structure (EXAFS) spectroscopy. J. Colloid Interface Sci. 2015, 442, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Newville, M. Fundamentals of XAFS. Rev. Miner. Geochem. 2014, 78, 33–74. [Google Scholar] [CrossRef]
- Rehr, J.J.; Albers, R.C. Theoretical approaches to x-ray absorption fine structure. Rev. Mod. Phys. 2000, 72, 621–654. [Google Scholar] [CrossRef]
- Trevorah, R.M.; Chantler, C.T.; Schalken, M.J. Solving self-absorption in fluorescence. IUCrJ 2019, 6, 1–17. [Google Scholar] [CrossRef]
- Tröger, L.; Arvanitis, D.; Baberschke, K.; Michaelis, H.; Grimm, U.; Zschech, E. Full correction of the self-absorption in soft-fluorescence extended x-ray-absorption fine structure. Phys. Rev. B 1992, 46, 3283–3289. [Google Scholar] [CrossRef]
- Booth, C.H.; Bridges, F. Improved SelfAbsorption Correction for Fluorescence Measurements of Extended XRay Absorption FineStructure. Phys. Scr. 2005, 2005, 202. [Google Scholar] [CrossRef]
- Goulon, J.; Goulon-Ginet, C.; Cortes, R.; Dubois, J.M. On experimental attenuation factors of the amplitude of the EXAFS oscillations in absorption, reflectivity and luminescence measurements. J. Phys. 1982, 43, 539–548. [Google Scholar] [CrossRef]
- Kelly, S.D.; Hesterberg, D.; Ravel, B. Analysis of Soils and Minerals Using X-ray Absorption Spectroscopy. In Methods of Soil Analysis Part 5—Mineralogical Methods; Ulery, A.L., Drees, L.R., Eds.; Soil Science Society of America: Madison, WI, USA; pp. 387–464. ISBN 978-0-89118-857-5.
- Meitzner, G.D.; Fischer, D.A. Distortions of fluorescence yield X-ray absorption spectra due to sample thickness. Microchem. J. 2002, 71, 281–286. [Google Scholar] [CrossRef]
- Hesterberg, D. Macroscale Chemical Properties and X-Ray Absorption Spectroscopy of Soil Phosphorus; Elsevier Masson SAS: Amsterdam, The Netherlands, 2010; Volume 34, ISBN 9780444532619. [Google Scholar]
- Prietzel, J.; Harrington, G.; Häusler, W.; Heister, K.; Werner, F.; Klysubun, W. Reference spectra of important adsorbed organic and inorganic phosphate binding forms for soil P speciation using synchrotron-based K-edge XANES spectroscopy. J. Synchrotron Radiat. 2016, 23, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Werner, F.; Prietzel, J. Standard Protocol and Quality Assessment of Soil Phosphorus Speciation by P K-Edge XANES Spectroscopy. Environ. Sci. Technol. 2015, 49, 10521–10528. [Google Scholar] [CrossRef] [PubMed]
- Ajiboye, B.; Akinremi, O.O.; Hu, Y.; Flaten, D.N. Phosphorus Speciation of Sequential Extracts of Organic Amendments Using Nuclear Magnetic Resonance and X-ray Absorption Near-Edge Structure Spectroscopies. J. Environ. Qual. 2007, 36, 1563. [Google Scholar] [CrossRef]
- Toor, G.S.; Hunger, S.; Peak, J.D.; Sims, J.T.; Sparks, D.L. Advances in the Characterization of Phosphorus in Organic Wastes: Environmental and Agronomic Applications. Adv. Agron. 2006, 89, 1–72. [Google Scholar]
- Pierzynski, G.M.; Logan, T.J.; Traina, S.J.; Bigham, J.M. Phosphorus Chemistry and Mineralogy in Excessively Fertilized Soils: Descriptions of Phosphorus-rich Particles. Soil Sci. Soc. Am. J. 1990, 54, 1583. [Google Scholar] [CrossRef]
- Ajiboye, B.; Akinremi, O.O.; Hu, Y.; Jürgensen, A. XANES Speciation of Phosphorus in Organically Amended and Fertilized Vertisol and Mollisol. Soil Sci. Soc. Am. J. 2008, 72, 1256. [Google Scholar] [CrossRef]
- Siebers, N.; Kruse, J.; Leinweber, P. Speciation of phosphorus and cadmium in a contaminated soil amended with bone char: Sequential fractionations and XANES spectroscopy. Water. Air. Soil Pollut. 2013, 224, 1564. [Google Scholar] [CrossRef]
- Prietzel, J.; Prater, I.; Colocho Hurtarte, L.C.; Hrbáček, F.; Klysubun, W.; Mueller, C.W. Site conditions and vegetation determine phosphorus and sulfur speciation in soils of Antarctica. Geochim. Cosmochim. Acta 2019, 246, 339–362. [Google Scholar] [CrossRef]
- Kruse, J.; Abraham, M.; Amelung, W.; Baum, C.; Bol, R.; Kühn, O.; Lewandowski, H.; Niederberger, J.; Oelmann, Y.; Rüger, C.; et al. Innovative methods in soil phosphorus research: A review. J. Plant Nutr. Soil Sci. 2015, 178, 43–88. [Google Scholar] [CrossRef] [Green Version]
- Ajiboye, B.; Akinremi, O.O.; Jürgensen, A. Experimental Validation of Quantitative XANES Analysis for Phosphorus Speciation. Soil Sci. Soc. Am. J. 2007, 71, 1288. [Google Scholar] [CrossRef]
- Alvarez, R.; Evans, L.A.; Milham, P.J.; Wilson, M.A. Effects of humic material on the precipitation of calcium phosphate. Geoderma 2004, 118, 245–260. [Google Scholar] [CrossRef]
- Güngör, K.; Jürgensen, A.; Karthikeyan, K.G. Determination of Phosphorus Speciation in Dairy Manure using XRD and XANES Spectroscopy. J. Environ. Qual. 2007, 36, 1856. [Google Scholar] [CrossRef]
- de Queiroz Paiva, A. Solos Carbonático-Fosfáticos do Platô de Irecê, BA: Genese, Mineralogia e Geoquimica; Universidade Federal de Vicosa: Viçosa-MG, Brazil, 2010. [Google Scholar]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Demirkiran, H.; Hu, Y.; Zuin, L.; Appathurai, N.; Aswath, P.B. XANES analysis of calcium and sodium phosphates and silicates and hydroxyapatite-Bioglass®45S5 co-sintered bioceramics. Mater. Sci. Eng. C 2011, 31, 134–143. [Google Scholar] [CrossRef]
- Sato, S.; Neves, E.G.; Solomon, D.; Liang, B.; Lehmann, J. Biogenic calcium phosphate transformation in soils over millennial time scales. J. Soils Sediments 2009, 9, 194–205. [Google Scholar] [CrossRef]
- Khare, N.; Hesterberg, D.; Beauchemin, S.; Wang, S.-L. XANES Determination of Adsorbed Phosphate Distribution between Ferrihydrite and Boehmite in Mixtures. Soil Sci. Soc. Am. J. 2004, 68, 460. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2018. Available online: https://www.r-bloggers.com/its-easy-to-cite-and-reference-r/ (accessed on 6 September 2019).
- Leri, A.C.; Hay, M.B.; Lanzirotti, A.; Rao, W.; Myneni, S.C.B. Quantitative determination of absolute organohalogen concentrations in environmental samples by X-ray absorption spectroscopy. Anal. Chem. 2006, 78, 5711–5718. [Google Scholar] [CrossRef] [PubMed]
- Le Guillou, C.; Bernard, S.; De La Pena, F.; Le Brech, Y. XANES-Based Quantification of Carbon Functional Group Concentrations. Anal. Chem. 2018, 90, 8379–8386. [Google Scholar] [CrossRef] [PubMed]
- Calvin, S. XAFS for Everyone; CRC Press Taylor and Francis: Boca Raton, FL, USA, 20 May 2013; ISBN 9781439878644. [Google Scholar]
- Khare, N.; Martin, J.D.; Hesterberg, D. Phosphate bonding configuration on ferrihydrite based on molecular orbital calculations and XANES fingerprinting. Geochim. Cosmochim. Acta 2007, 71, 4405–4415. [Google Scholar] [CrossRef]
- Beauchemin, S.; Hesterberg, D.; Chou, J.; Beauchemin, M.; Simard, R.R.; Sayers, D.E. Speciation of Phosphorus in Phosphorus-Enriched Agricultural Soils Using X-Ray Absorption Near-Edge Structure Spectroscopy and Chemical Fractionation. J. Environ. Qual. 2003, 32, 1809. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, Y.; Yang, J.; Abdi, D.; Cade-Menun, B.J. Investigation of Soil Legacy Phosphorus Transformation in Long-Term Agricultural Fields Using Sequential Fractionation, P K-edge XANES and Solution P NMR Spectroscopy. Environ. Sci. Technol. 2015, 49, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Bauke, S.L.; von Sperber, C.; Siebers, N.; Tamburini, F.; Amelung, W. Biopore effects on phosphorus biogeochemistry in subsoils. Soil Biol. Biochem. 2017, 111, 157–165. [Google Scholar] [CrossRef]
- Egger, M.; Jilbert, T.; Behrends, T.; Rivard, C.; Slomp, C.P. Vivianite is a major sink for phosphorus in methanogenic coastal surface sediments. Geochim. Cosmochim. Acta 2015, 169, 217–235. [Google Scholar] [CrossRef] [Green Version]
- Seiter, J.M.; Staats-Borda, K.E.; Ginder-Vogel, M.; Sparks, D.L. XANES Spectroscopic Analysis of Phosphorus Speciation in Alum-Amended Poultry Litter. J. Environ. Qual. 2008, 37, 477. [Google Scholar] [CrossRef]
- Xu, X.; Thornton, P.E.; Post, W.M. A global analysis of soil microbial biomass carbon, nitrogen and phosphorus in terrestrial ecosystems. Glob. Ecol. Biogeogr. 2013, 22, 737–749. [Google Scholar] [CrossRef]
- Klysubun, W.; Tarawarakarn, P.; Thamsanong, N.; Amonpattaratkit, P.; Cholsuk, C.; Lapboonrueng, S.; Chaichuay, S.; Wongtepa, W. Upgrade of SLRI BL8 beamline for XAFS spectroscopy in a photon energy range of 1–13 keV. Radiat. Phys. Chem. 2019. [Google Scholar] [CrossRef]
- Petric, M.; Bohinc, R.; Bučar, K.; Žitnik, M.; Szlachetko, J.; Kavčič, M. Chemical state analysis of phosphorus performed by X-ray emission spectroscopy. Anal. Chem. 2015, 87, 5632–5639. [Google Scholar] [CrossRef] [PubMed]

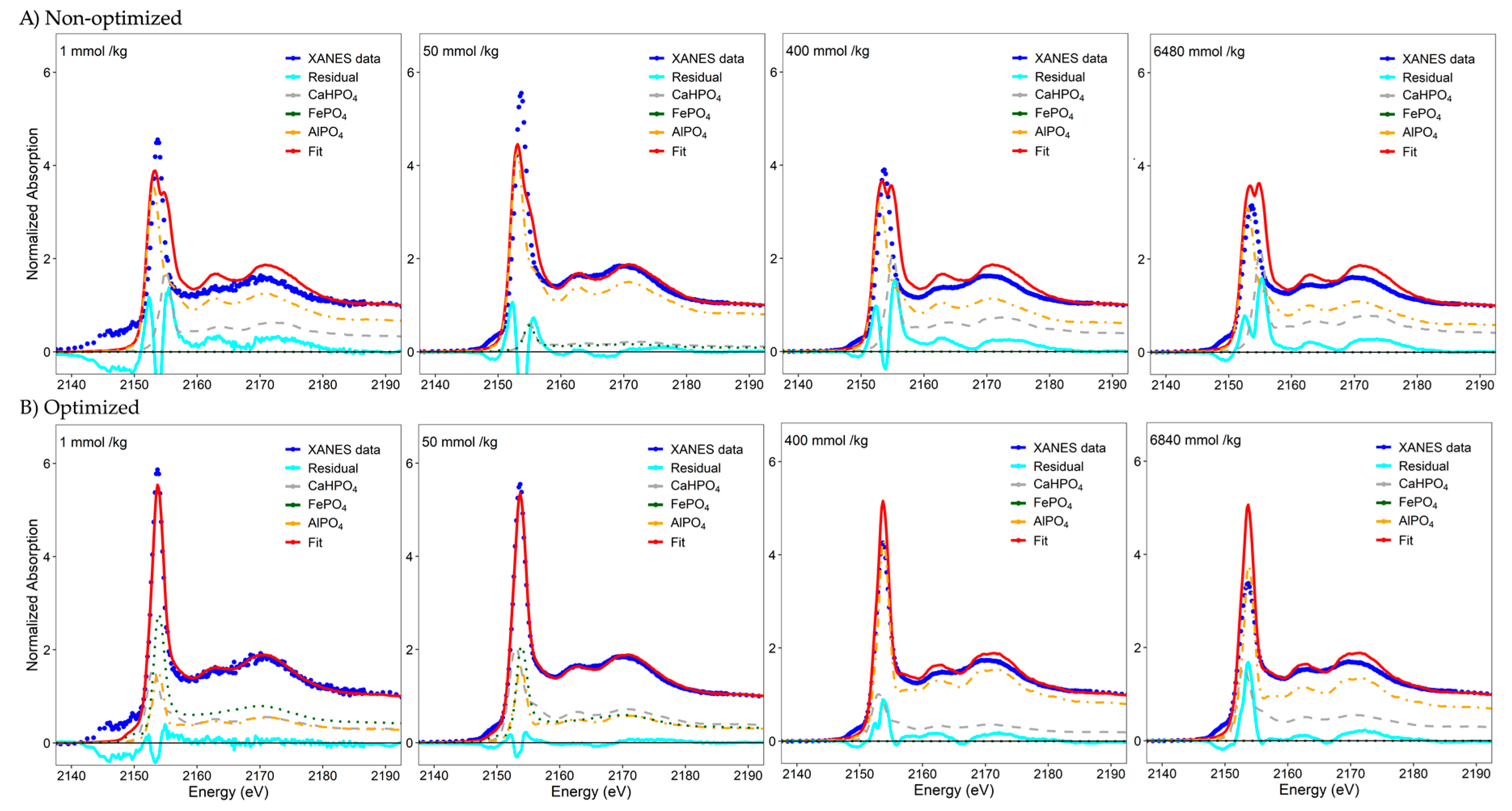
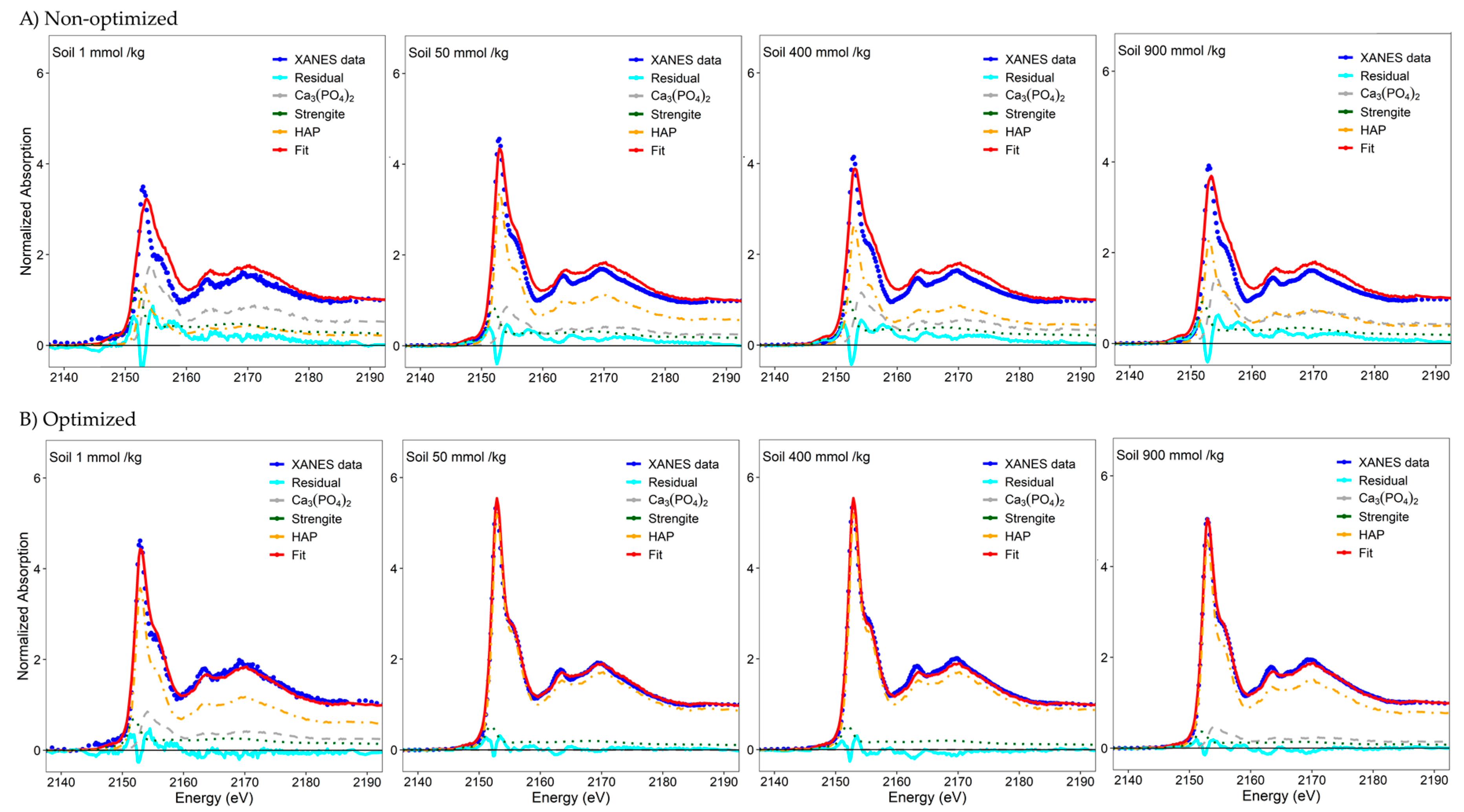


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P Content (mmol kg−1) | Non-Optimized Values | Relative Deviation from Total P | R-Factor | Optimized Values | Relative Deviation from Total P | R-Factor | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FeP * | AlP * | CaP * | FeP | AlP | CaP | FeP | AlP | CaP | FeP | AlP | CaP | |||
| % | % | |||||||||||||
| 1 | 0 | 83 | 17 | −33 | +50 | −16 | 0.0283 | 42 | 29 | 30 | +9 | −4 | −3 | 0.0067 |
| 6.25 | 19 | 53 | 27 | −14 | +20 | −6 | 0.0044 | 42 | 24 | 34 | +9 | −9 | +1 | 0.0030 |
| 12.5 | 57 | 0 | 43 | +24 | −33 | +10 | 0.0196 | 29 | 31 | 39 | −4 | −2 | +6 | 0.0041 |
| 25 | 53 | 0 | 47 | +20 | −33 | +14 | 0.0048 | 34 | 24 | 42 | +1 | −9 | +9 | 0.0031 |
| 50 | 31 | 31 | 38 | −2 | −2 | +5 | 0.0016 | 31 | 31 | 38 | −2 | −2 | +5 | 0.0016 |
| 75 | 32 | 34 | 34 | −1 | +1 | +1 | 0.0012 | 33 | 33 | 34 | 0 | 0 | +1 | 0.0012 |
| 100 | 6 | 69 | 25 | −27 | +36 | −8 | 0.0020 | 25 | 44 | 31 | −8 | +11 | −2 | 0.0009 |
| 300 | 0 | 74 | 26 | −33 | +41 | −7 | 0.0036 | 18 | 48 | 34 | −15 | +15 | +1 | 0.0011 |
| 400 | 0 | 85 | 15 | −33 | +52 | −18 | 0.0357 | 0 | 81 | 19 | −33 | +48 | −14 | 0.0132 |
| 1000 | 0 | 76 | 24 | −33 | +43 | −9 | 0.0159 | 0 | 71 | 29 | −33 | +38 | −4 | 0.0041 |
| 6840 | 0 | 75 | 25 | −33 | +42 | −8 | 0.0703 | 0 | 71 | 29 | −33 | +38 | −4 | 0.0435 |
| Mean: | 18 | 53 | 29 | 23 | 44 | 33 | ||||||||
| SD: | 22 | 32 | 10 | 16 | 21 | 6 | ||||||||
| P Content (mmol kg−1) | Non-Optimized Values | R-Factor | Optimized Values | R-Factor | ||||
|---|---|---|---|---|---|---|---|---|
| TCP * | Apatite | Strengite | TCP * | Apatite | Strengite | |||
| % | % | |||||||
| 1 | 51 | 23 | 27 | 0.038283 | 24 | 61 | 14 | 0.004991 |
| 6.25 | 28 | 58 | 13 | 0.013889 | 0 | 94 | 6 | 0.001992 |
| 12.5 | 22 | 66 | 12 | 0.007424 | 0 | 100 | 0 | 0.000994 |
| 25 | 17 | 74 | 9 | 0.005962 | 0 | 100 | 0 | 0.000927 |
| 50 | 24 | 58 | 18 | 0.014383 | 0 | 89 | 11 | 0.001857 |
| 75 | 40 | 41 | 19 | 0.022008 | 17 | 73 | 9 | 0.001701 |
| 100 | 37 | 46 | 17 | 0.020679 | 18 | 74 | 8 | 0.002005 |
| 200 | 32 | 48 | 20 | 0.023714 | 0 | 89 | 11 | 0.001978 |
| 300 | 39 | 44 | 17 | 0.020381 | 19 | 73 | 8 | 0.001511 |
| 400 | 33 | 45 | 22 | 0.023067 | 0 | 89 | 11 | 0.001858 |
| 900 | 42 | 39 | 19 | 0.027448 | 14 | 78 | 8 | 0.001190 |
| Mean: | 33 | 49 | 17 | 8 | 83 | 8 | ||
| SD: | 10 | 14 | 5 | 10 | 11 | 4 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colocho Hurtarte, L.C.; Souza-Filho, L.F.; Oliveira Santos, W.; Vergütz, L.; Prietzel, J.; Hesterberg, D. Optimization of Data Processing Minimizes Impact of Self-Absorption on Phosphorus Speciation Results by P K-Edge XANES. Soil Syst. 2019, 3, 61. https://doi.org/10.3390/soilsystems3030061
Colocho Hurtarte LC, Souza-Filho LF, Oliveira Santos W, Vergütz L, Prietzel J, Hesterberg D. Optimization of Data Processing Minimizes Impact of Self-Absorption on Phosphorus Speciation Results by P K-Edge XANES. Soil Systems. 2019; 3(3):61. https://doi.org/10.3390/soilsystems3030061
Chicago/Turabian StyleColocho Hurtarte, Luis Carlos, Luiz Francisco Souza-Filho, Wedisson Oliveira Santos, Leonardus Vergütz, Jörg Prietzel, and Dean Hesterberg. 2019. "Optimization of Data Processing Minimizes Impact of Self-Absorption on Phosphorus Speciation Results by P K-Edge XANES" Soil Systems 3, no. 3: 61. https://doi.org/10.3390/soilsystems3030061