Sputtered Platinum Thin-films for Oxygen Reduction in Gas Diffusion Electrodes: A Model System for Studies under Realistic Reaction Conditions

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Pt Thin-Film Electrodes
2.2. Physical Characterization
2.3. Chemicals, Materials and Gases
2.4. Gas Diffusion Electrode (GDE)
2.5. Electrochemical Measurements
3. Results and Discussion
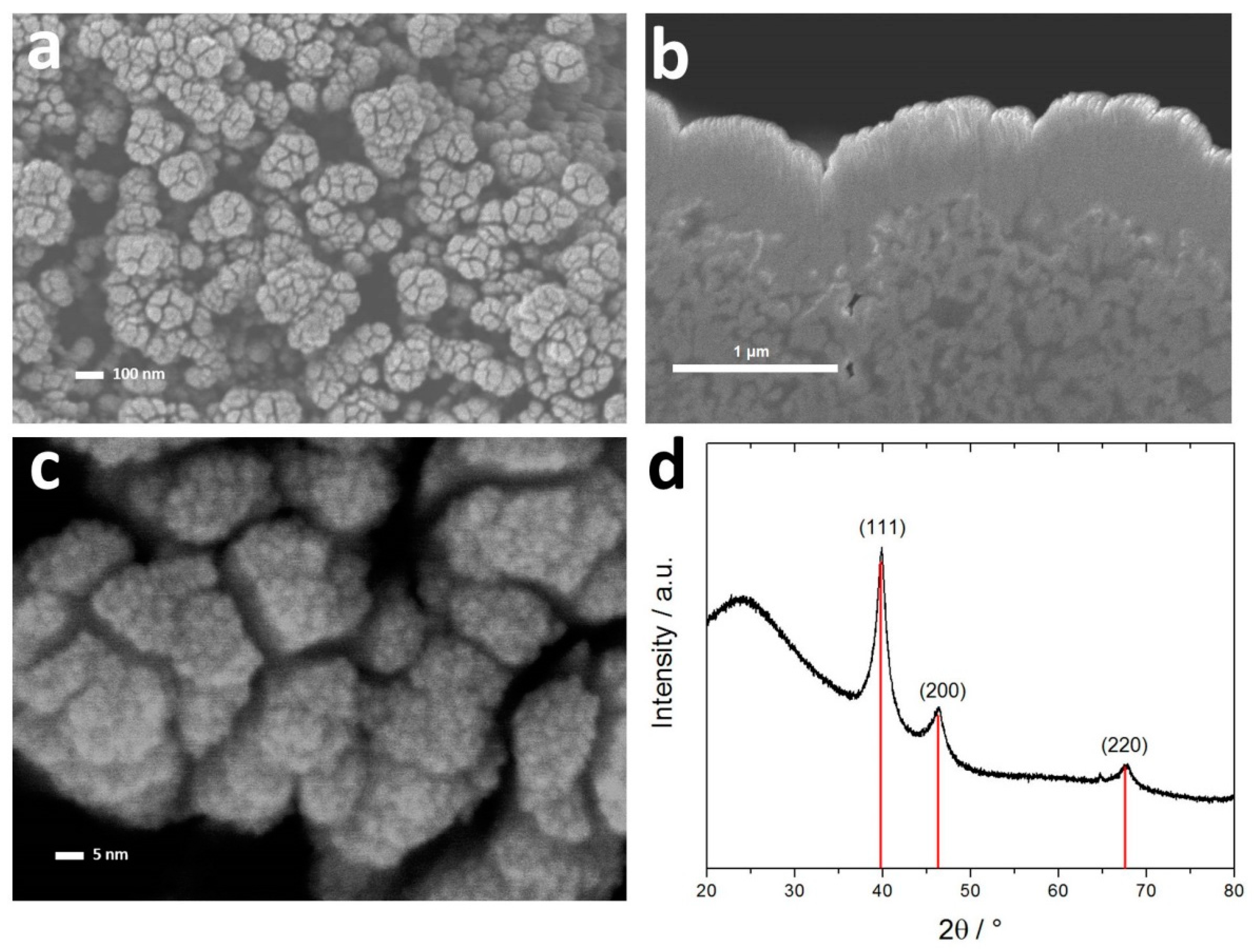
3.1. Structural Characterization of Pt Thin-Films
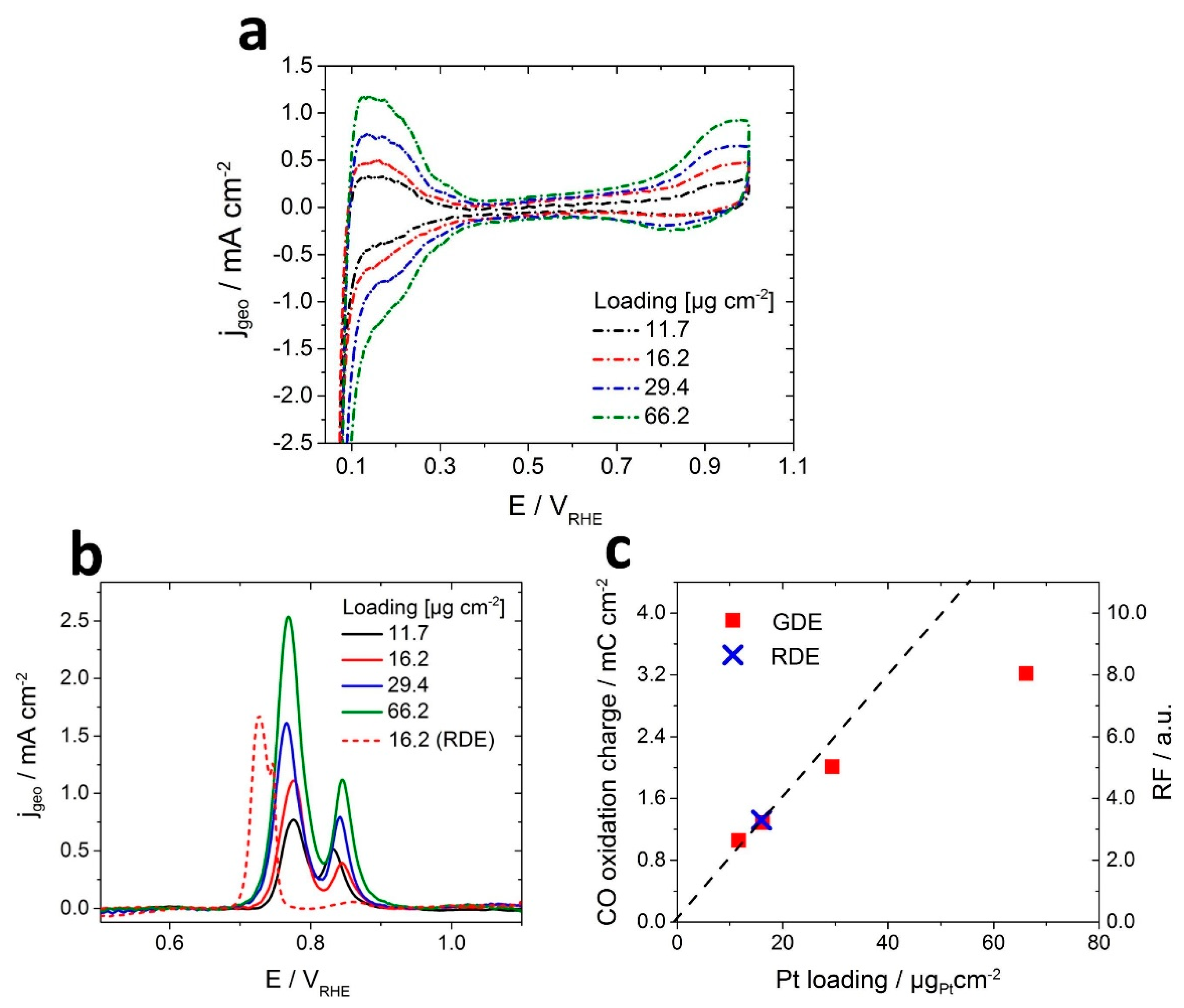
3.2. Electrochemical Characterization
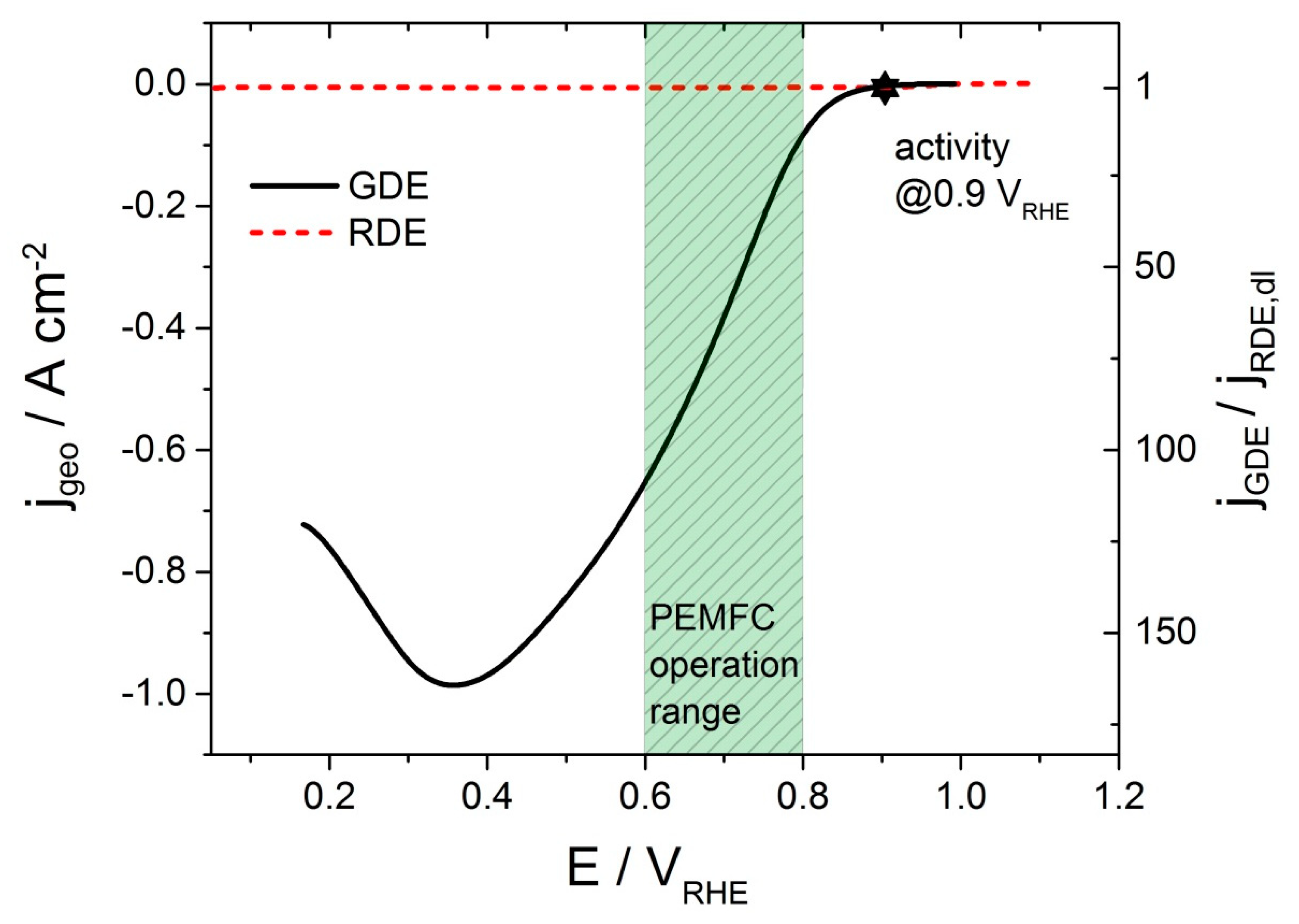
3.3. Performance of Pt Thin-Films in the GDE Setup
3.4. Benchmarking Different Pt Loadings in the GDE Setup
3.5. Systematic Change of Operation Parameters
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nat. Cell Biol. 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Rabis, A.; Rodriguez, P.; Schmidt, T.J. Electrocatalysis for Polymer Electrolyte Fuel Cells: Recent Achievements and Future Challenges. ACS Catal. 2012, 2, 864–890. [Google Scholar] [CrossRef]
- Wagner, F.T.; Lakshmanan, B.; Mathias, M.F. Electrochemistry and the Future of the Automobile. J. Phys. Chem. Lett. 2010, 1, 2204–2219. [Google Scholar] [CrossRef]
- Mathias, M.F.; Kongkanand, A. The Priority and Challenge of High-Power Performance of Low-Platinum Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. Lett. 2016, 7, 1127–1137. [Google Scholar]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Marković, N.M. Improved Oxygen Reduction Activity on Pt3Ni(111) via Increased Surface Site Availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Banham, D.; Ye, S. Current Status and Future Development of Catalyst Materials and Catalyst Layers for Proton Exchange Membrane Fuel Cells: An Industrial Perspective. ACS Energy Lett. 2017, 2, 629–638. [Google Scholar] [CrossRef]
- Strmcnik, D.; Escudero-Escribano, M.; Kodama, K.; Stamenkovic, V.R.; Cuesta, A.; Marković, N.M. Enhanced electrocatalysis of the oxygen reduction reaction based on patterning of platinum surfaces with cyanide. Nat. Chem. 2010, 2, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Stamenković, V.; Mun, B.S.; Mayrhofer, K.J.J.; Ross, P.N.; Marković, N.M.; Rossmeisl, J.; Greeley, J.; Nørskov, J.K. Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure. Angew. Chem. Int. Ed. 2006, 45, 2897–2901. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Jensen, K.D.; Jensen, A.W. Recent advances in bimetallic electrocatalysts for oxygen reduction: Design principles, structure-function relations and active phase elucidation. Curr. Opin. Electrochem. 2018, 8, 135–146. [Google Scholar] [CrossRef]
- Stamenkovic, V.R.; Mun, B.S.; Arenz, M.; Mayrhofer, K.J.J.; Lucas, C.A.; Wang, G.; Ross, P.N.; Marković, N.M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241–247. [Google Scholar] [CrossRef]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar] [CrossRef]
- Escudero-Escribano, M.; Malacrida, P.; Hansen, M.H.; Vej-Hansen, U.G.; Velázquez-Palenzuela, A.; Tripkovic, V.; Schiøtz, J.; Rossmeisl, J.; Stephens, I.E.L.; Chorkendorff, I. Tuning the activity of Pt alloy electrocatalysts by means of the lanthanide contraction. Science 2016, 352, 73–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, I.E.L.; Rossmeisl, J.; Chorkendorff, I. Toward sustainable fuel cells. Science 2016, 354, 1378–1379. [Google Scholar] [CrossRef]
- Han, B.; Kukreja, R.S.; Theobald, B.R.; O’Malley, R.; Wagner, F.T.; Carlton, C.E.; Kongkanand, A.; Gan, L.; Strasser, P.; Shao-Horn, Y. Record activity and stability of dealloyed bimetallic catalysts for proton exchange membrane fuel cells. Energy Environ. Sci. 2015, 8, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Mayrhofer, K.; Strmcnik, D.; Blizanac, B.; Stamenković, V.; Arenz, M.; Marković, N. Measurement of oxygen reduction activities via the rotating disc electrode method: From Pt model surfaces to carbon-supported high surface area catalysts. Electrochim. Acta 2008, 53, 3181–3188. [Google Scholar] [CrossRef]
- Pedersen, C.M.; Escudero-Escribano, M.; Velázquez-Palenzuela, A.; Christensen, L.H.; Chorkendorff, I.; Stephens, I.E. Benchmarking Pt-based electrocatalysts for low temperature fuel cell reactions with the rotating disk electrode: Oxygen reduction and hydrogen oxidation in the presence of CO (review article). Electrochim. Acta 2015, 179, 647–657. [Google Scholar] [CrossRef]
- Zamburlini, E.; Jensen, K.D.; Stephens, I.E.; Chorkendorff, I.; Escudero-Escribano, M. Benchmarking Pt and Pt-lanthanide sputtered thin films for oxygen electroreduction: Fabrication and rotating disk electrode measurements. Electrochim. Acta 2017, 247, 708–721. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B: Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Gasteiger, H.; Panels, J.; Yan, S. Dependence of PEM fuel cell performance on catalyst loading. J. Power Sources 2004, 127, 162–171. [Google Scholar] [CrossRef]
- O’Hayre, R.; Prinz, F.B. The Air/Platinum/Nafion Triple-Phase Boundary: Characteristics, Scaling, and Implications for Fuel Cells. J. Electrochem. Soc. 2004, 151, A756. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Schmutz, P.; Frankel, G.S. Characterization of High-Surface-Area Electrocatalysts Using a Rotating Disk Electrode Configuration. J. Electrochem. Soc. 1998, 145, 2354. [Google Scholar] [CrossRef]
- Zana, A.; Wiberg, G.K.H.; Deng, Y.-J.; Østergaard, T.; Rossmeisl, J.; Arenz, M. Accessing the Inaccessible: Analyzing the Oxygen Reduction Reaction in the Diffusion Limit. ACS Appl. Mater. Interfaces 2017, 9, 38176–38180. [Google Scholar] [CrossRef]
- Zalitis, C.M.; Kramer, D.; Kucernak, A.R. Electrocatalytic performance of fuel cell reactions at low catalyst loading and high mass transport. Phys. Chem. Chem. Phys. 2013, 15, 4329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Jiang, Y.; Jensen, J.O.; Cleemann, L.N.; Li, Q. Catalyst evaluation for oxygen reduction reaction in concentrated phosphoric acid at elevated temperatures. J. Power Sour. 2018, 375, 77–81. [Google Scholar] [CrossRef]
- Wiberg, G.K.H.; Fleige, M.; Arenz, M. Gas diffusion electrode setup for catalyst testing in concentrated phosphoric acid at elevated temperatures. Sci. Instrum. 2015, 86, 024102. [Google Scholar] [CrossRef]
- Pinaud, B.A.; Bonakdarpour, A.; Daniel, L.; Sharman, J.; Wilkinson, D.P. Key Considerations for High Current Fuel Cell Catalyst Testing in an Electrochemical Half-Cell. J. Electrochem. Soc. 2017, 164, F321–F327. [Google Scholar] [CrossRef]
- Inaba, M.; Jensen, A.W.; Sievers, G.W.; Escudero-Escribano, M.; Zana, A.; Arenz, M. Benchmarking high surface area electrocatalysts in a gas diffusion electrode: measurement of oxygen reduction activities under realistic conditions. Energy Environ. Sci. 2018, 11, 988–994. [Google Scholar] [CrossRef]
- Jouny, M.; Luc, W.; Jiao, F. High-rate electroreduction of carbon monoxide to multi-carbon products. Nat. Catal. 2018, 1, 748–755. [Google Scholar] [CrossRef]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; De Arquer, F.P.G.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Möller, T.; Ju, W.; Bagger, A.; Wang, X.; Luo, F.; Thanh, T.N.; Varela, A.S.; Rossmeisl, J.; Strasser, P.; Moeller, T. Efficient CO2 to CO electrolysis on solid Ni–N–C catalysts at industrial current densities. Energy Environ. Sci. 2019, 12, 640–647. [Google Scholar] [CrossRef]
- Higgins, D.; Hahn, C.; Xiang, C.; Jaramillo, T.F.; Weber, A.Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm. ACS Energy Lett. 2019, 4, 317–324. [Google Scholar] [CrossRef]
- Chen, C.; Kang, Y.; Huo, Z.; Zhu, Z.; Huang, W.; Xin, H.L.; Snyder, J.D.; Li, D.; Herron, J.A.; Mavrikakis, M.; et al. Highly Crystalline Multimetallic Nanoframes with Three-Dimensional Electrocatalytic Surfaces. Science 2014, 343, 1339–1343. [Google Scholar] [CrossRef]
- Sepa, D.; Vojnovic, M.; Damjanovic, A. Reaction intermediates as a controlling factor in the kinetics and mechanism of oxygen reduction at platinum electrodes. Electrochim. Acta 1981, 26, 781–793. [Google Scholar] [CrossRef]
- Garcia-Araez, N.; Climent, V.; Feliu, J. Potential-Dependent Water Orientation on Pt(111), Pt(100), and Pt(110), As Inferred from Laser-Pulsed Experiments. Electrostatic and Chemical Effects. J. Phys. Chem. C 2009, 113, 9290–9304. [Google Scholar] [CrossRef]
- Láng, G.; Horanyi, G. Some interesting aspects of the catalytic and electrocatalytic reduction of perchlorate ions. J. Electroanal. Chem. 2003, 552, 197–211. [Google Scholar] [CrossRef]
- McBreen, J. Voltammetric Studies of Electrodes in Contact with Ionomeric Membranes. J. Electrochem. Soc. 1985, 132, 1112. [Google Scholar] [CrossRef]
- Liu, J.; Zenyuk, I.V. Proton transport in ionomer-free regions of polymer electrolyte fuel cells and implications for oxygen reduction reaction. Curr. Opin. Electrochem. 2018, 12, 202–208. [Google Scholar] [CrossRef]
- Debe, M.K. Tutorial on the Fundamental Characteristics and Practical Properties of Nanostructured Thin Film (NSTF) Catalysts. J. Electrochem. Soc. 2013, 160, F522–F534. [Google Scholar] [CrossRef]
- Nesselberger, M.; Ashton, S.; Meier, J.C.; Katsounaros, I.; Mayrhofer, K.J.J.; Arenz, M. The Particle Size Effect on the Oxygen Reduction Reaction Activity of Pt Catalysts: Influence of Electrolyte and Relation to Single Crystal Models. J. Am. Chem. Soc. 2011, 133, 17428–17433. [Google Scholar] [CrossRef] [PubMed]
- Inaba, M.; Suzuki, T.; Hatanaka, T.; Morimoto, Y. Fabrication and Cell Analysis of a Pt/SiO2 Platinum Thin Film Electrode. J. Electrochem. Soc. 2015, 162, F634–F638. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Layer (Reference) | Electrolyte | Loading [µgPt cm−2] | Temperature/Humidity [°C] | SA @0.9VRHE [mA cm−2Pt] | SA @0.65VRHE [mA cm−2Pt] | ECSA [m2 g−1Pt] |
|---|---|---|---|---|---|---|
| Pt TF RDE (this work) | 0.1 M HClO4 | 16 | rt | 1.82 | - | 21 |
| Pt TF GDE (this work) | Nafion 117/4 M HClO4 | 16 | rt/0% RH | 0.72 | 144 | 20 |
| Pt TF GDE (this work) | 4 M HClO4 | 16 | rt/0% RH | 0.71 | 67.5 | 20 |
| Pt/C RDE [40] | 0.1 M HClO4 | 14 | rt | 0.49 | - | 76 |
| Pt/C GDE [28] | Nafion 117/4 M HClO4 | 5 | rt/100% RH | 0.14 | 25.9 | 93 |
| Pt/C GDE [28] | Nafion 117/4 M HClO4 | 5 | 60/100% RH | 0.18 | 47.9 | 81 |
| Pt/C MEA [41] | Nafion 117 | 90 | 80/100% RH | 0.21 | - | 80 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sievers, G.W.; Jensen, A.W.; Brüser, V.; Arenz, M.; Escudero-Escribano, M. Sputtered Platinum Thin-films for Oxygen Reduction in Gas Diffusion Electrodes: A Model System for Studies under Realistic Reaction Conditions. Surfaces 2019, 2, 336-348. https://doi.org/10.3390/surfaces2020025
Sievers GW, Jensen AW, Brüser V, Arenz M, Escudero-Escribano M. Sputtered Platinum Thin-films for Oxygen Reduction in Gas Diffusion Electrodes: A Model System for Studies under Realistic Reaction Conditions. Surfaces. 2019; 2(2):336-348. https://doi.org/10.3390/surfaces2020025
Chicago/Turabian StyleSievers, Gustav W., Anders W. Jensen, Volker Brüser, Matthias Arenz, and María Escudero-Escribano. 2019. "Sputtered Platinum Thin-films for Oxygen Reduction in Gas Diffusion Electrodes: A Model System for Studies under Realistic Reaction Conditions" Surfaces 2, no. 2: 336-348. https://doi.org/10.3390/surfaces2020025