

DFT and MCDS Outcome for a Comparative Analysis of NO, NO2, SO, SO2 and SO3 Gas Adsorption onto a NaMgPO4 (033) Surface

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Quantum Chemical Calculations
2.2. Monte Carlo Dynamic Simulation Details
3. Results
3.1. Frontier Molecular Orbitals and MEP
3.2. Global Quantum Descriptors
3.3. Mulliken Charge Distribution
3.4. Fukui Function Calculations
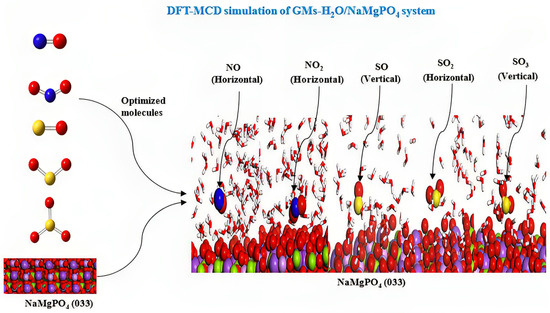
3.5. Monte Carlo Dynamic Simulation Study
3.6. Radial Distribution Function
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, L.; Zhong, Z.; Yang, H.; Wang, C.; Wang, L. DeNOx performance and characteristic study for transition metals doped iron-based catalysts. Korean J. Chem. Eng. 2017, 34, 1229. [Google Scholar] [CrossRef]
- Shahzad, K.; Saleem, M.; Ghauri, M.; Akhtar, J.; Ali, N.; Akhtar, N.A. Combustion Science and 6th Asian Physics Symposium. IOP Publ. J. Phy. Conf. Ser. 2015, 187, 1079–1092. [Google Scholar]
- Nugraha; Saputro, A.G.; Agusta, M.K.; Yuliarto, B.; Dipojono, H.K.; Maezono, R. Density functional study of adsorptions of CO2, NO2 and SO2 molecules on Zn(0002) surfaces. J. Phys. Conf. Ser. 2016, 739, 012080. [Google Scholar] [CrossRef]
- Streets, D.; Waldhoff, S. Present and future emissions of air pollutants in China: SO2, NOx, and CO. Atmos. Environ. 2000, 34, 363–374. [Google Scholar] [CrossRef]
- Sakai, Y.; Koyanagi, M.; Mogi, K.; Miyoshi, E. Theoretical study of adsorption of SO2 on Ni(111) and Cu(111) surfaces. Surf. Sci. 2002, 513, 272–282. [Google Scholar] [CrossRef]
- Lin, F.; Wu, X.; Liu, S.; Weng, D.; Huang, Y. Preparation of MnOx-CeO2–Al2O3 mixed oxides for NOx-assisted soot oxidation: Activity, structure and thermal stability. J. Chem. Eng. 2013, 226, 105–112. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Y.B.; He, H. Oxygen vacancies on nanosized ceria govern the NOx storage capacity of NSR catalysts. Catal. Sci. Technol. 2016, 6, 3950–3962. [Google Scholar] [CrossRef]
- Rezaei, F.; Rownaghi, A.A.; Monjezi, S.; Lively, R.P.; Jones, C.W. SOx/NOx removal from flue gas streams by solid adsorbents: A review of current challenges and future directions. Energy Fuels 2015, 29, 5467–5486. [Google Scholar] [CrossRef]
- Liu, L.; Gao, X.; Song, H.; Zheng, C.H.; Zhu, X.B.; Luo, Z.Y.; Ni, M.J.; Cen, K.F. Study of the promotion effect of iron on supported manganese catalysts for NO oxidation. Aerosol. Air Qual. Res. 2014, 14, 1038–1046. [Google Scholar] [CrossRef]
- Guo, R.T.; Chen, Q.L.; Ding, H.L.; Wang, Q.S.; Pan, W.G.; Yang, N.Z.; Lu, C.Z. Preparation and characterization of CeOx@MnOx core-shell structure catalyst for catalytic oxidation of NO. Catal. Commun. 2015, 69, 165–169. [Google Scholar] [CrossRef]
- Zahaf, R.; Jung, J.W.; Coker, Z.; Kim, S.; Choi, T.Y.; Lee, D. Pt catalyst over SiO2 and Al2O3 supports synthesized by aerosol method for HC-SCR DeNOx application. Aerosol. Air Qual. Res. 2015, 15, 2409–2421. [Google Scholar] [CrossRef]
- Chen, T.; Lin, H.; Guan, B.; Gong, X.; Li, K.; Huang, Z. Promoting the low temperature activity of Ti–V–O catalysts by premixed flame synthesis. J. Chem. Eng. 2016, 296, 45–55. [Google Scholar] [CrossRef]
- Guo, R.T.; Sun, P.; Pan, W.G.; Li, M.Y.; Liu, S.M.; Sun, X.; Liu, S.W.; Liu, J. A highly effective MnNdOx catalyst for the selective catalytic reduction of NOx with NH3. Ind. Eng. Chem. Res. 2017, 56, 12566–12577. [Google Scholar] [CrossRef]
- Granger, P.; Parvulescu, V.I. Catalytic NOx Abatement Systems for Mobile Sources: From Three-Way to Lean Burn after-Treatment Technologies. Chem. Rev. 2011, 111, 3155–3207. [Google Scholar] [CrossRef]
- Feng, H.; Wang, C.; Huang, Y. Particle deposition behaviors of monolithic De-NOx catalysts for selective catalytic reduction (SCR). Korean J. Chem. Eng. 2017, 34, 2832–2839. [Google Scholar] [CrossRef]
- Jackson, G.J.; Driver, S.M.; Woodruff, D.P.; Abrams, N.; Jones, R.G.; Butterfield, M.T.; Rapper, M.D.C.; Cowied, B.C.C.; Formoso, V. A structural study of the interaction of SO2 with Cu(111). Surf. Sci. 2000, 459, 231–244. [Google Scholar] [CrossRef]
- Terada, S.; Yokoyama, T.; Sakano, M.; Kiguchi, M.; Kitajima, Y.; Ohta, T. Asymmetric surface structure of SO2 on Pd(111) studied by total-reflection X-ray absorption fine structure spectroscopy. Chem. Phys. Lett. 1999, 300, 645–650. [Google Scholar] [CrossRef]
- Wilson, K.; Hardacre, C.; Baddeley, C.J.; Ludecke, J.; Woodruff, D.P.; Lsmbert, R.M. A spectroscopic study of the chemistry and reactivity of SO2 on Pt{111}: Reactions with O2, CO and C3H6. Surf. Sci. 1997, 372, 279–288. [Google Scholar] [CrossRef]
- Harrison, M.J.; Woodruff, D.P.; Robinson, J. Density functional theory investigation of the structure of SO2 and SO3 on Cu(111) and Ni(111). Surf. Sci. 2006, 600, 1827–1836. [Google Scholar] [CrossRef]
- Wanglai, C.; Meiling, H.; Jie, L.; Shandong, Y.; Yongjun, L.; Yinghao, C. Oxidation of SO2 and NO by epoxy groups on graphene oxides: The role of the hydroxyl group. RSC Adv. 2015, 5, 22802–22810. [Google Scholar]
- Oladele, E.O.; Alabi, A.H.; Olawale, M.D.; Ishaya, F.A. Adsorption of methylene blue dye from stimulated wastewater onto modified and unmodified cassis fistula pods: Kinetics, thermodynamics and equilibrium studies. UNIOSUN J. Sci. 2019, 4, 1–14. [Google Scholar]
- Monticelli, O.; Loenders, R.; Jacobs, P.A.; Martens, J.A. NOx removal from exhaust gas from lean burn internal combustion engines through adsorption on FAU type zeolites cation exchanged with alkali metals and alkaline earth metals. Appl. Catal. B Environ. 1999, 21, 215–220. [Google Scholar] [CrossRef]
- Li, L.; Chen, J.; Zhang, S.; Guan, N.; Wang, T.; Liu, S. Selective catalytic reduction of nitrogen oxides from exhaust of lean burn engine over in situ synthesized monolithic Cu–TS-1/cordierite. Catal. Today 2004, 90, 207–213. [Google Scholar] [CrossRef]
- Sultana, A.; Habermacher, D.D.; Kirschhock, C.E.A.; Martens, J.A. Adsorptive separation of NOx in presence of SOx from gas mixtures simulating lean burn engine exhaust by pressure swing process on Na-Y zeolite. Appl. Catal. B Environ. 2004, 48, 65–76. [Google Scholar] [CrossRef]
- Barka, N.; Assabbane, A.; Nounahb, A.; Laanab, L.; Ichou, Y.A. Removal of textile dyes from aqueous solutions by natural phosphate as a new adsorbent. Desalination 2009, 235, 264–275. [Google Scholar] [CrossRef]
- Sajid, H.; Siddique, S.A.; Ahmed, E.; Arshad, M.; Gilani, M.A.; Rauf, A.; Imran, M.; Mahmood, T. DFT outcome for comparative analysis of Be12O12, Mg12O12 and Ca12O12 nanocages toward sensing of N2O, NO2, NO, H2S, SO2 and SO3 gases. Comput. Theor. Chem. 2022, 1211, 113694. [Google Scholar] [CrossRef]
- Gao, Z.; Li, L.; Huang, H.; Xu, S.; Yan, G.; Zhao, M.; Ding, Z. Adsorption characteristics of acid gases (NO, NO2, SO2 and SO3) on different single-atom nickel adsorbent: A first-principles study. Appl. Surf. Sci. 2020, 527, 146939. [Google Scholar] [CrossRef]
- Siddique, S.A.; Sajid, H.; Gilani, M.A.; Ahmed, E.; Arshad, M.; Mahmood, T. Sensing of SO3, SO2, H2S, NO2 and N2O toxic gases through aza-macrocycle via DFT calculations. Comput. Theor. Chem. 2022, 1209, 113606. [Google Scholar] [CrossRef]
- Balakrishna, A.; Ntwaeaborwa, O.M. Study of luminescent behavior and crystal defects of different MNa[PO4]-Dy3+ phosphors (M = Mg, Ca, Sr and Ba). Sens. Actuators B Chem. 2017, 242, 305–317. [Google Scholar] [CrossRef]
- Bhavya, N.R.; Mahendra, M.; Doreswamy, B.H.; Kumar, S.; Gilandoust, M.; El-khatatneh, N.A. Computational and spectroscopic investigations on boronic acid based fluorescent carbohydrate sensor in aqueous solution at physiological pH 7. 5. J. Mol. Struct. 2019, 1194, 305–319. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Shokuhi Rad, A.; Zardoost, M.R.; Abedini, E. First-principles study of terpyrrole as a potential hydrogen cyanide sensor: DFT calculations. J. Mol. Model. 2015, 21, 273. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical hardness and density functional theory. J. Chem. Sci. 2005, 117, 369–377. [Google Scholar] [CrossRef]
- Geerling, P.; Prof, F.D.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G.; Szentpaly, L.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar]
- Kaya, S.; Guo, L.; Kaya, C.; Tüzün, B.; Obot, I.B.; Touir, R.; Islam, N. Quantum chemical and molecular dynamic simulation studies for the prediction of inhibition efeciencies of some piperidine derivatives on the corrosion of iron, J. Taiwan Inst. Chem. Eng. 2016, 65, 522–529. [Google Scholar] [CrossRef]
- BIOVIA Materials Studio Version 8.0; Accelrys Inc.: San Diego, CA, USA, 2016.
- Khnifira, M.; El Hamidi, S.; Sadiq, M.; Şimşek, S.; Kaya, S.; Barka, N.; Abdennouri, M. Adsorption mechanisms investigation of methylene blue on the (001) zeolite 4A surface in aqueous medium by computational approach and molecular dynamics. Appl. Surf. Sci. 2022, 572, 151381. [Google Scholar] [CrossRef]
- Hsissou, R.; Benhiba, F.; Abbout, S.; Dagdag, O.; Benkhaya, S.; Berisha, A.; Erramli, H.; Elharfi, A. Trifunctional epoxy polymer as corrosion inhibition material for carbon steel in 1.0 M HCl: MD simulations, DFT and complexation computations. Inorg. Chem. Commun. 2020, 115, 107858. [Google Scholar] [CrossRef]
- Ajebli, S.; Kaichouh, G.; Khachani, M.; Babas, H.; El Karbane, M.; Warad, I.; Safi, Z.S.; Berisha, A.; Mehmeti, V.; Guenbour, A.; et al. The adsorption of Tenofovir in aqueous solution on activated carbon produced from maize cobs: Insights from experimental, molecular dynamics simulation, and DFT calculations. Chem. Phys. Lett. 2022, 801, 139676. [Google Scholar] [CrossRef]
- Khnifira, M.; El Hamidi, S.; Machrouhi, A.; Mahsoune, A.; Boumya, W.; Tounsadi, H.; Mahjoubi, F.Z.; Sadiq, M.; Barka, N.; Abdennouri, M. Theoretical and experimental study of the adsorption characteristics of Methylene Blue on titanium dioxide surface using DFT and Monte Carlo dynamic simulation. Desalin. Water Treat. 2020, 190, 393–411. [Google Scholar] [CrossRef]
- Kokalj, A. Molecular modeling of organic corrosion inhibitors: Calculations, pitfalls, and conceptualization of molecule–surface bonding. Corros. Sci. 2021, 193, 109650. [Google Scholar] [CrossRef]
- Vengatesh, G.; Sundaravadivelu, M. Experimental and theoretical evaluation of new piperidine and oxaquinuclidine core containing derivatives as an efficient corrosion inhibitor for copper in nitric acid medium. J. Adhes. Sci. Technol. 2020, 34, 2075–2106. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Wang, H.; Wang, L.; Liu, A. DFT study of new bipyrazole derivatives and their potential activity as corrosion inhibitors. J. Mol. Model. 2007, 13, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Sastri, V.S.; Perumareddi, J.R. Molecular Orbital Theoretical Studies of Some Organic Corrosion Inhibitors. Corros. Sci. 1997, 53, 617–622. [Google Scholar] [CrossRef]
- Abdulazeez, M.O.; Oyebamiji, A.K.; Semire, B. DFT and QSAR study of corrosion inhibition on 3,5-di-substituted pyrazol derivatives with heteroatom on position one. Leban. Sci. J. 2016, 17, 217–232. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, Y.; Shu, X.; Wang, Y.; Ran, Q. Adsorption of organic molecules on mineral surfaces studied by first-principle calculations: A review. Adv. Colloid Interface Sci. 2018, 256, 230–241. [Google Scholar] [CrossRef]
- Khnifira, M.; Mahsoune, A.; Belghiti, M.E.; Khamar, L.; Sadiq, M.; Abdennouri, M.; Barka, N. Combined DFT and MD simulation approach for the study of SO2 and CO2 adsorption on graphite (111) surface in aqueous medium. CRGSC Curr. Res. Green Sustain. Chem. 2021, 4, 100085. [Google Scholar] [CrossRef]
- Kondori, J.; Zendehboudi, S.; James, L. Molecular dynamic simulations to evaluate dissociation of hydrate structure II in the presence of inhibitors: A mechanistic study, Chem. Eng. Res. Des. 2019, 149, 81–94. [Google Scholar] [CrossRef]
- Khnifira, M.; Boumya, W.; Attarki, J.; Mahsoune, A.; Abdennouri, M.; Sadiq, M.; Kaya, S.; Barka, N. Elucidating the adsorption mechanisms of anionic dyes on chitosan (110) surface in aqueous medium by quantum chemical and molecular dynamics. Mater. Today Commun. 2022, 33, 104488. [Google Scholar] [CrossRef]
- Khnifira, M.; Boumya, W.; Attarki, J.; Soufi, A.; Sadiq, M.; Achak, M.; Barka, N.; Abdennouri, M. Interaction between drug molecule and inverse spinel surfaces in aqueous solution: Insights from DFT and DMC simulation. Comput. Theor. Chem. 2023, 1228, 114289. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (eV) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Molecule | EHOMO | ELUMO | Eg | S | χ | η | µ | ω | Eb–d | ΔN |
| NO | −9.434 | −3.236 | 6.198 | 0.323 | 6.335 | 3.099 | −6.335 | −1.584 | −0.775 | 1.022 |
| NO2 | −9.766 | −9.403 | 0.363 | 5.510 | 9.584 | 0.181 | −9.584 | −2.396 | −0.045 | 26.403 |
| SO | −8.123 | −5.297 | 2.825 | 0.708 | 6.710 | 1.412 | −6.710 | −1.677 | −0.353 | 2.375 |
| SO2 | −9.100 | −6.461 | 2.639 | 0.758 | 7.781 | 1.320 | −7.781 | −1.945 | −0.330 | 2.948 |
| SO3 | −12.193 | −10.318 | 1.874 | 1.067 | 11.255 | 0.937 | −11.255 | −2.814 | −0.234 | 6.004 |
| N | O | S | |
|---|---|---|---|
| NO | 0.7797 | −0.7797 | |
| NO2 | 0.2779 | −0.1389 | |
| SO | −0.5456 | 0.5456 | |
| SO2 | −0.5216 | 1.0432 | |
| SO3 | −0.4687 −0.4535 −0.4531 | 1.3753 |
| Molecule | Atom | qi(N) | qi(N + 1) | qi(N − 1) | fi+ | fi− | Δf |
|---|---|---|---|---|---|---|---|
| NO | O1 | −0.202 | 0.214 | −0.565 | 0.416 | 0.363 | 0.053 |
| N2 | 0.202 | 0.786 | −0.435 | 0.584 | 0.637 | −0.053 | |
| NO2 | N1 | 0.498 | 0.194 | 0.837 | −0.304 | −0.339 | 0.035 |
| O2 | −0.249 | 0.081 | −0.597 | 0.33 | 0.348 | −0.018 | |
| O3 | −0.249 | 0.081 | −0.597 | 0.33 | 0.348 | −0.018 | |
| SO | S1 | 0.584 | 1.374 | −0.228 | 0.79 | 0.812 | −0.022 |
| O2 | −0.584 | −0.374 | −0.772 | 0.21 | 0.188 | 0.022 | |
| SO2 | S1 | 1.423 | 1.684 | 0.84 | 0.261 | 0.583 | −0.322 |
| O2 | −0.711 | −0.342 | −0.92 | 0.369 | 0.209 | 0.16 | |
| O3 | −0.711 | −0.342 | −0.92 | 0.369 | 0.209 | 0.16 | |
| SO3 | S1 | 1.985 | 2.025 | 1.427 | 0.04 | 0.558 | −0.518 |
| O2 | −0.662 | −0.339 | −0.809 | 0.323 | 0.147 | 0.176 | |
| O3 | −0.662 | −0.341 | −0.809 | 0.321 | 0.147 | 0.174 | |
| O4 | −0.661 | −0.345 | −0.809 | 0.316 | 0.148 | 0.168 |
| Molecule | Etotal | Eads | RAE | Edef | dEads/dNiH2O | dEads/dNi |
|---|---|---|---|---|---|---|
| NO | −472.031 | −874.033 | −472.563 | −374.469 | −0.764 | −2.218 |
| NO2 | −433.835 | −819.943 | −434.982 | −384.961 | −0.781 | −13.714 |
| SO | −481.969 | −924.810 | −482.146 | −442.664 | −0.766 | −72.933 |
| SO2 | −474.448 | −876.333 | −475.027 | −401.306 | −0.769 | −31.010 |
| SO3 | −447.572 | −977.714 | −453.463 | −524.251 | −0.804 | −155.016 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attarki, J.; Khnifira, M.; Boumya, W.; Hajjaoui, H.; Mahsoune, A.; Sadiq, M.; Achak, M.; Barka, N.; Abdennouri, M. DFT and MCDS Outcome for a Comparative Analysis of NO, NO2, SO, SO2 and SO3 Gas Adsorption onto a NaMgPO4 (033) Surface. Surfaces 2023, 6, 450-465. https://doi.org/10.3390/surfaces6040030
Attarki J, Khnifira M, Boumya W, Hajjaoui H, Mahsoune A, Sadiq M, Achak M, Barka N, Abdennouri M. DFT and MCDS Outcome for a Comparative Analysis of NO, NO2, SO, SO2 and SO3 Gas Adsorption onto a NaMgPO4 (033) Surface. Surfaces. 2023; 6(4):450-465. https://doi.org/10.3390/surfaces6040030
Chicago/Turabian StyleAttarki, Jamal, Malika Khnifira, Wafaa Boumya, Hind Hajjaoui, Anass Mahsoune, M’hamed Sadiq, Mounia Achak, Noureddine Barka, and Mohamed Abdennouri. 2023. "DFT and MCDS Outcome for a Comparative Analysis of NO, NO2, SO, SO2 and SO3 Gas Adsorption onto a NaMgPO4 (033) Surface" Surfaces 6, no. 4: 450-465. https://doi.org/10.3390/surfaces6040030