
Unveiling the Origin of the Selectivity and the Molecular Mechanism in the [3+2] Cycloaddition Reaction of N-aryl-C-carbamoylnitrone with N-arylitaconimide


Abstract
:1. Introduction
2. Computational Methods
- -
- The Gaussian 09 [33] was used for the optimization of all the stationery points, namely, the reagents, transition states and cycloadducts.
- -
- -
- The frequency calculations were used to confirm that the optimized stationery points were TSs or minimums. The structure of the TSs were used to confirm that the mechanism was one step or stepwise by performing intrinsic reaction coordinate (IRC) calculations [37], in which the obtained curve indicates that the TS structure is connected to the two minimums, in the forward direction to the cycloadduct and in the reverse direction to the separated reagents.
- -
- -
- The values of thermochemistry properties such as Gibbs free energies, enthalpies and entropies were calculated using standard statistical thermodynamics at 298.15 K and 1 atm through the optimized gas phase structures [42].
- -
- The electrophilicity index (ω) [43], which is a measure of the molecule’s ability to accept electrons, is given by the relation ω = (μ2/2η).
- -
- The electronic chemical potential (μ) and the chemical hardness (η) were calculated directly from the energies of the frontier molecular orbitals εHOMO and εLUMO, respectively, by the following relations: μ = (εHOMO + εLUMO)/2 and η = εLUMO − εHOMO [44].
- -
- The nucleophilic index [45], (N), which is a measure of the molecular power of the donating electrons, is given by the relation N = εHOMO(Nu) − εHOMO(TCE), in which TCE is tetracyanoethylene, which is taken as the reference due to its low HOMO energy.
- -
- The Parr functions [46], namely, the electrophilic index () and nucleophilic one (), which allowed us to characterize the most electrophilic and nucleophilic atoms in a molecule, were obtained by an analysis of the Mulliken atomic spin density of the radical anion and the radical cation, respectively, of the studied molecule.
- -
- The dual reactivity descriptors were used in order to determine, atthe same time, the nucleophilic and the electrophilic atomic region in the studied molecule. These descriptors were obtained from the difference between nucleophilic and electrophilic Fukui functions [47] as follow:where ρN+1 ®®(r) an®N−1(r) are the electronic densities at point r for a system with (N + 1), (N) and (N − 1) electrons, respectively.
- -
- The Wiberg bond indices were used to analyze the synchronicity of the mechanism [48].
- -
3. Results and Discussion
3.1. Analysis of Reactivity
3.1.1. Analysis of Regioselectivity
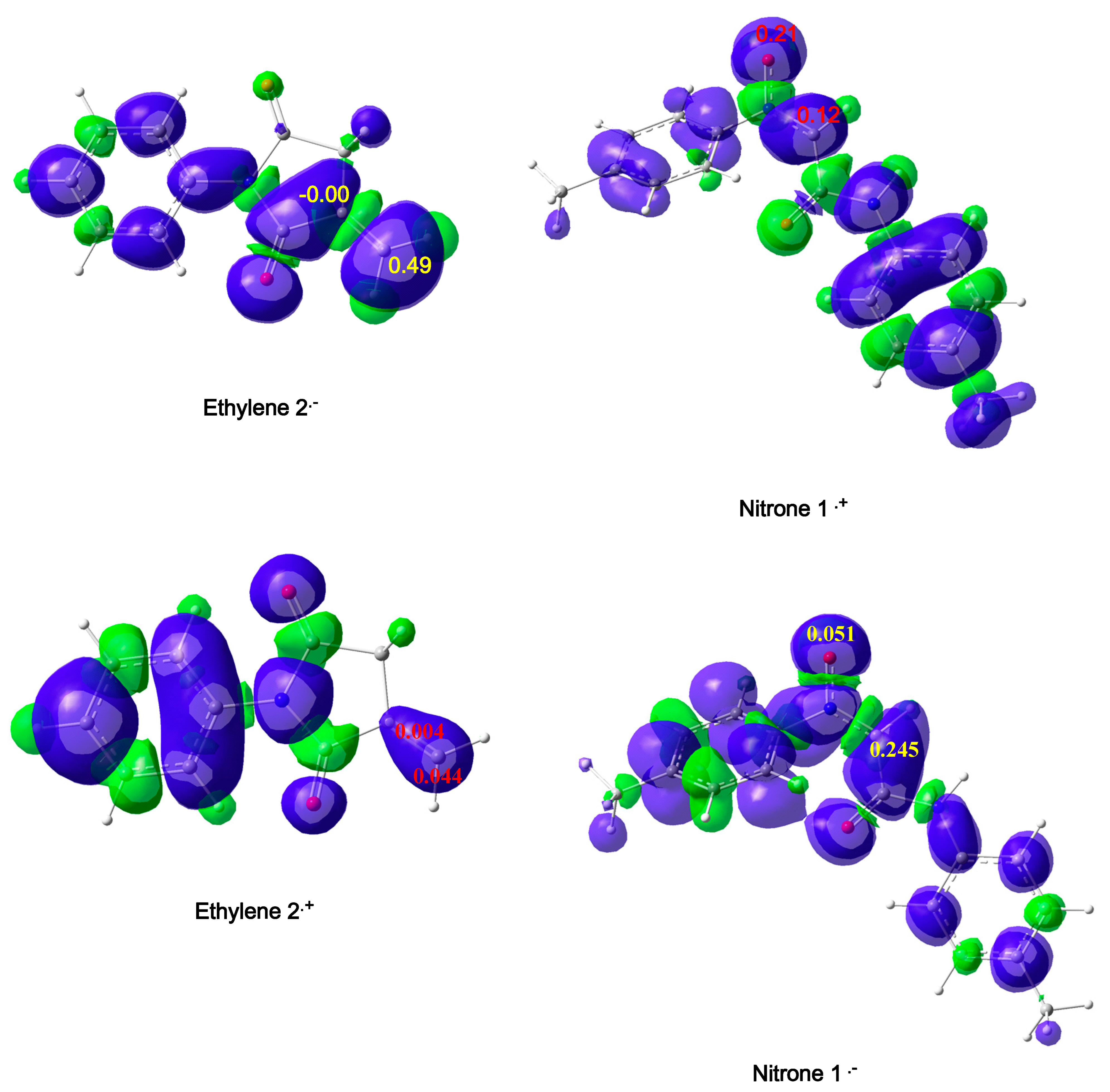
Using Parr Functions
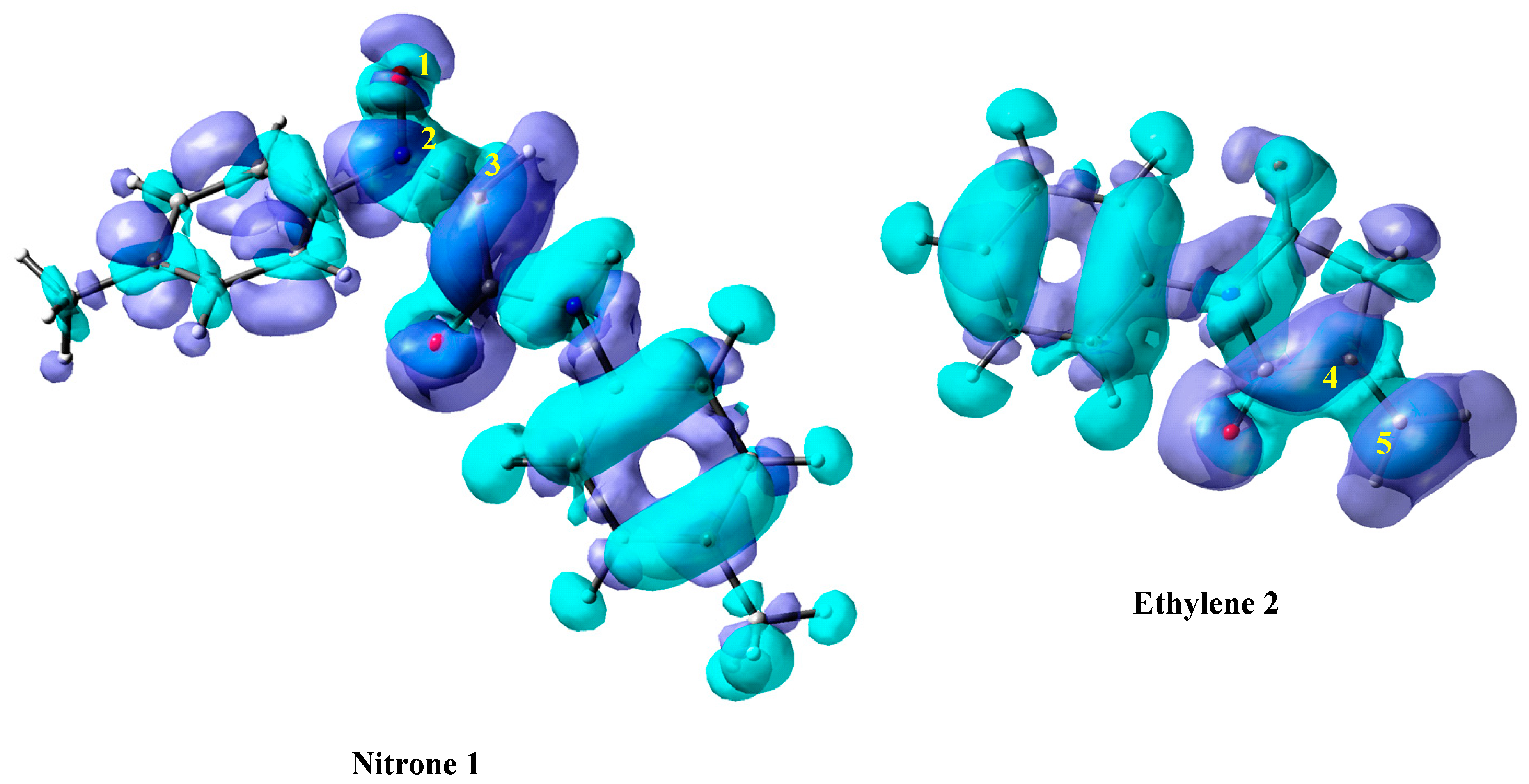
Using Dual Descriptors
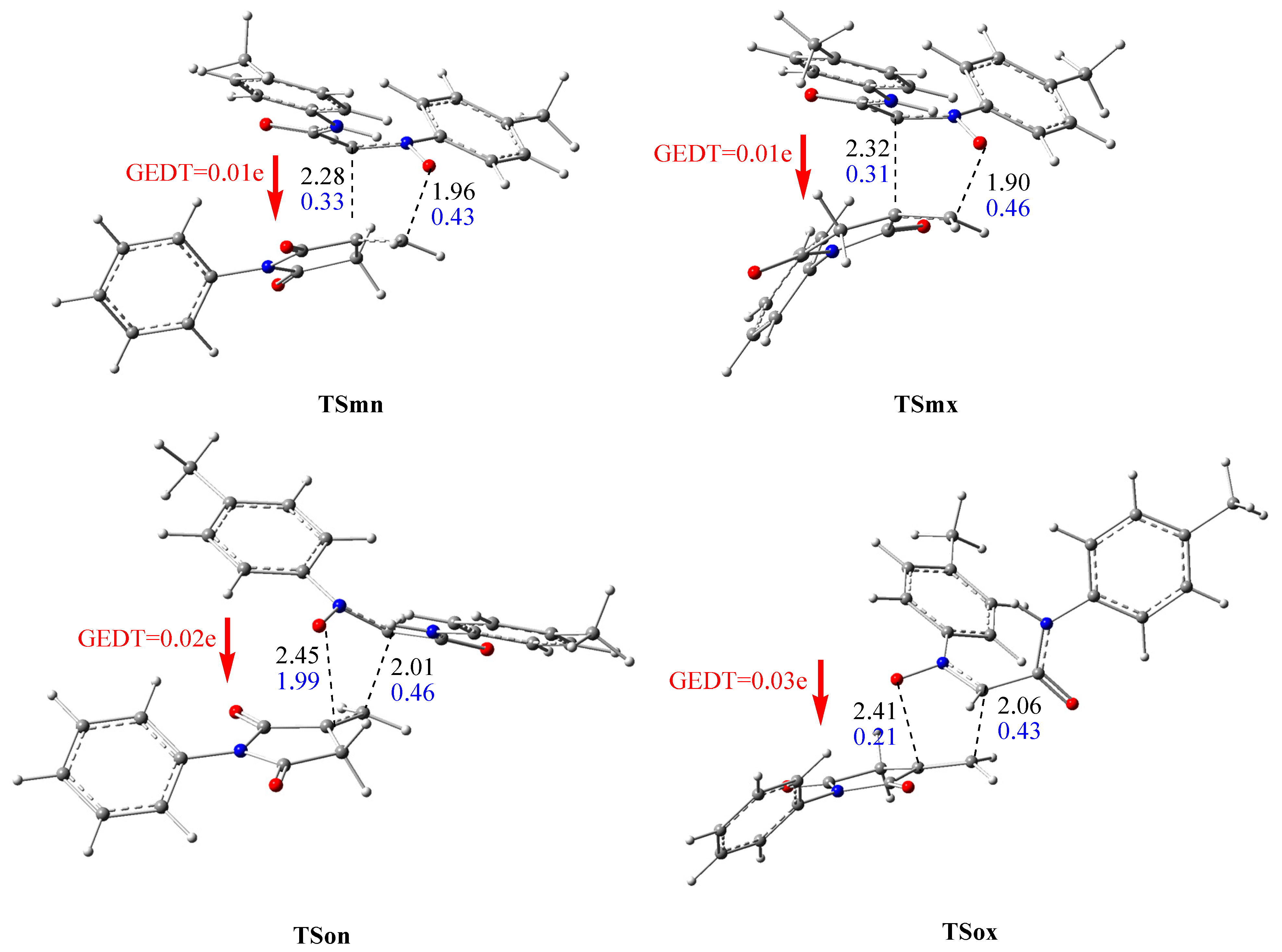
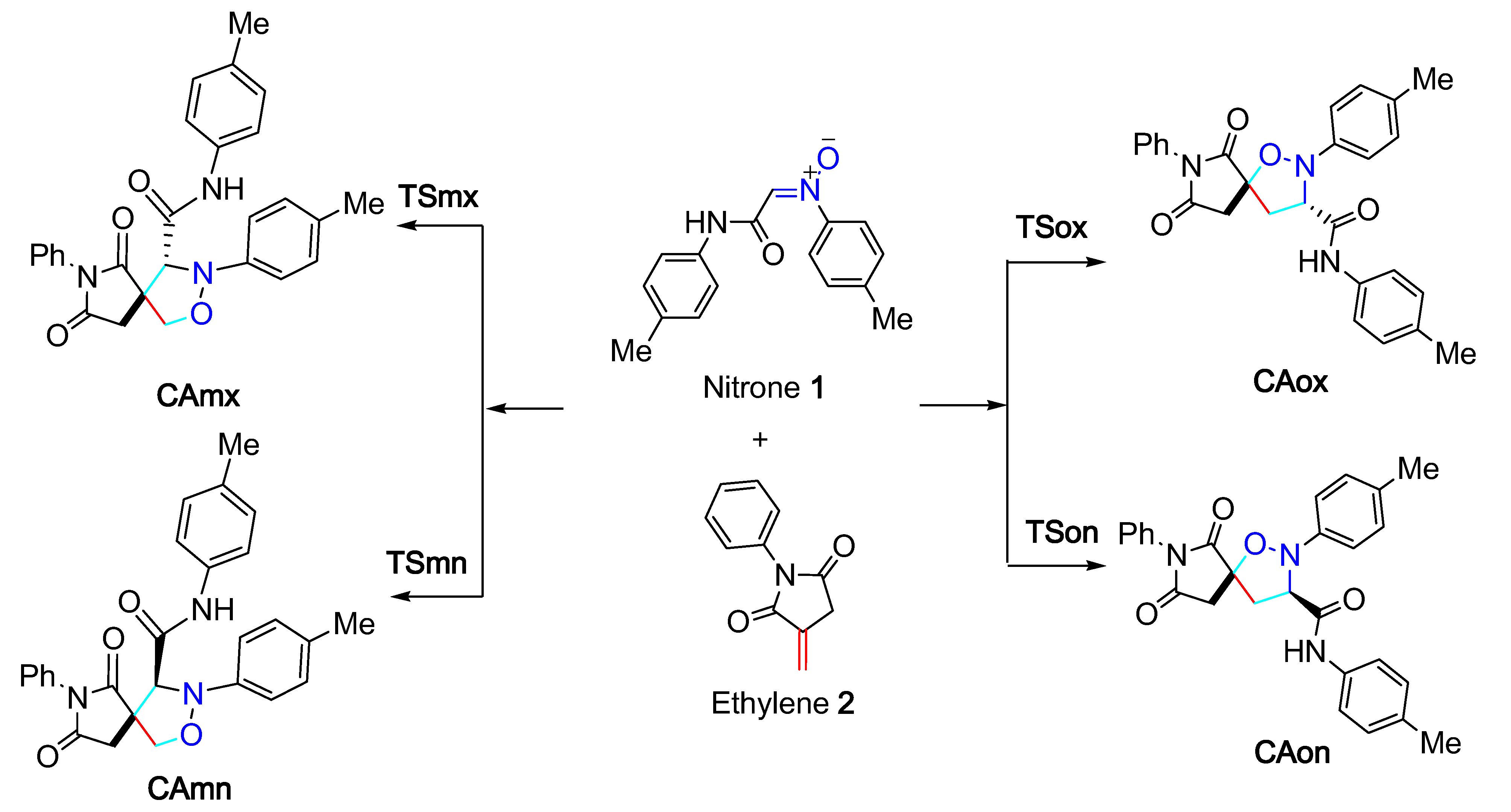
3.2. Geometries of TSs and Reaction Polarity Analysis
3.3. Energy Profiles Analysis
3.3.1. Gas Phase Electronic Energy Analysis
3.3.2. Solvent Effects
3.3.3. Thermochemistry Analysis
4. Conclusions
- The CDFT global reactivity indices explain the low polar character of this 32CA reaction, which is related to the close electronic parameters of the reagents.
- An analysis on reactivity and selectivity using the local indices obtained from the Parr functions explain well the experimentally ortho regioselectivity and indicates that nitrone 1 is an electrophile while ethylene 2 is a nucleophile.
- The analysis of regioselectivity using dual reactivity indices allows us to explain well the observed ortho regioselectivity.
- The analysis of the molecular mechanism of this 32CA reaction using the GEDT and Wiberg indices indicated that it is a non-polar, one-step, asynchronous mechanism for the favored ortho pathways and synchronous for the meta ones.
- The energy profile gas phase analysis showed that the studied 32CA reaction was characterized by an ortho regioselectivity and an endo stereoselectivity, in accordance with the experimental observation.
- The analysis of the solvent effects indicated that dichloromethane did not have any influence on the selectivity but it increased the activation energies and decreased the relative energies of the cycloadducts associated with this 32CA reaction.
- A thermodynamic parameters analysis showed that this 32CA was ortho regioselective and endo stereoselective, was under kinetic control and had exothermic and exergonic characters.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duarte, C.D.; Barreiro, E.J.; Fraga, C.A. Privileged structures: A useful concept for the rational design of new lead drug candidates. Mini-Rev. Med. Chem. 2007, 7, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Yar, M.S. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, H.; Zhou, J.; Wang, W.; He, Y.; Wang, C. Application of primary halogenated hydrocarbons for the synthesis of 3-aryl and 3-alkyl indolizines. Org. Biomol. Chem. 2017, 15, 5016–5024. [Google Scholar] [CrossRef] [PubMed]
- Zagórska, A.; Kołaczkowski, M.; Bucki, A.; Siwek, A.; Kazek, G.; Satała, G.; Bojarski, A.J.; Partyka, A.; Wesołowska. A Pawłowski, M. Structure–activity relationships and molecular studies of novel arylpiperazinylalkyl purine-2,4-diones and purine-2,4,8-triones with antidepressant and anxiolytic-like activity. Eur. J. Med. Chem. 2015, 97, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Kouadri, Y.; Ouahrani, M.R.; Missaoui, B.E.; Chebrouk, F.; Gherraf, N. Reactivity of b-Cetoesters Compounds, Synthesis of Nitrogenated Heterocycles (Derivatives of Tetrahydroacridin-9-ones and Pyrimidinone) and Biological Properties of Pyrimidinone Derivatives. Asian J. Chem. 2015, 27, 3675–3680. [Google Scholar] [CrossRef]
- Xie, M.; Lapidus, R.G.; Sadowska, M.; Edelman, M.J.; Hosmane, R.S. Synthesis, anticancer activity, and SAR analyses of compounds containing the 5: 7-fused 4, 6, 8-triaminoimidazo [4,5-e][1,3] diazepine ring system. Bioorg. Med. Chem. 2016, 24, 2595–2602. [Google Scholar] [CrossRef]
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Comp. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Patterson, J.W.; Cheung, P.S.; Ernest, M.J. 3-Carboxy-5-methyl-N-[4-(trifluoromethyl) phenyl]-4-isoxazolecarboxamide, new prodrug for the antiarthritic agent 2-cyano-3-hydroxy-N-[4-(trifluoromethyl) phenyl]-2-butenamide. J. Med. Chem. 1992, 35, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Becan, L.; Nowakowska, E. Synthesis and pharmacological assessment of derivatives of isoxazolo [4 5-d] pyrimidine. Bioorg. Med. Chem. 2004, 12, 265–272. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloaddition Chemistry; Wiley: New York, NY, USA, 1984; Volume 1, pp. 42–47. [Google Scholar]
- Huisgen, R. 1, 3-dipolar cycloadditions. Past and future. Angew. Chem. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Rios, R. Enantioselective methodologies for the synthesis of spiro compounds. Chem. Soc. Rev. 2012, 41, 1060–1074. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xie, Y.J.; Yan, C.G. Construction of dispirocyclopentanebisoxindoles via self-domino Michael-aldol reactions of 3-phenacylideneoxindoles. J. Org. Chem. 2013, 78, 8354–8365. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Xing, G.J.; Zhu, R.Y.; Tan, W.; Tu, S. A catalytic asymmetric isatin-involved povarov reaction: Diastereo-and enantioselective construction of spiro [indolin-3, 2′-quinoline] scaffold. Org. Lett. 2013, 15, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Khan, M.N.; Karamthulla, S.; Abbas, S.J.; Choudhury, L.H. One pot four-component reaction for the efficient synthesis of spiro [indoline-3,4′-pyrano [2,3-c] pyrazole]-3′-carboxylate derivatives. Tetrahedron Lett. 2013, 54, 5434–5440. [Google Scholar] [CrossRef]
- Rapposelli, S.; Breschi, M.C.; Calderone, V.; Digiacomo, M.; Martelli, A.; Testai, L.; Vanni, M.; Balsamo, A. Synthesis and biological evaluation of 5-membered spiro heterocycle-benzopyran derivatives against myocardial ischemia. Eur. J. Med. Chem. 2011, 46, 966–973. [Google Scholar] [CrossRef]
- Sadeghian, Z.; Bayat, M.; Safari, F. Synthesis and antitumor activity screening of spiro tryptanthrin-based heterocyclic compounds. Med. Chem. Res. 2022, 31, 497–506. [Google Scholar] [CrossRef]
- Cheng, G.J.; Zhang, X.; Chung, L.W.; Xu, L.; Wu, Y.D. Computational organic chemistry: Bridging theory and experiment in establishing the mechanisms of chemical reactions. J. Am. Chem. Soc. 2015, 137, 1706–1725. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Domingo, L.R. Unravelling the mysteries of the [3+2] cycloaddition reactions. Eur. J. Org. Chem. 2019, 2–3, 267–282. [Google Scholar] [CrossRef]
- Jørgensen, K.A. Cycloaddition Reactions in Organic Synthesis; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Chafaa, F.; Hellel, D.; Nacereddine, A.K.; Djerourou, A. A theoretical study of the regio-and stereoselectivities of non-polar 1, 3-dipolar cycloaddition reaction between C-diethoxyphosphoryl-N-methylnitrone and N-(2-fluorophenyl) acrylamide. Mol. Phys. 2016, 114, 663–670. [Google Scholar] [CrossRef]
- Chafaa, F.; Khorief Nacereddine, A.; Djerourou, A. Unravelling the mechanism and the origin of the selectivity of the [3+2] cycloaddition reaction between electrophilic nitrone and nucleophilic alkene. Theor. Chem. Acc. 2019, 138, 1–11. [Google Scholar] [CrossRef]
- Nacereddine, A.K.; Layeb, H.; Chafaa, F.; Yahia, W.; Djerourou, A.; Domingo, L.R. A DFT study of the role of the Lewis acid catalysts in the [3+2] cycloaddition reaction of the electrophilic nitrone isomer of methyl glyoxylate oxime with nucleophilic cyclopentene. RSC Adv. 2015, 5, 64098–64105. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A molecular electron density theory study of the reactivity and selectivities in [3+ 2] cycloaddition reactions of C, N-dialkyl nitrones with ethylene derivatives. J. Org. Chem. 2018, 83, 2182–2197. [Google Scholar] [CrossRef]
- Nacereddine, A.K.; Yahia, W.; Bouacha, S.; Djerourou, A. A theoretical investigation of the regio-and stereoselectivities of the 1, 3-dipolar cycloaddition of C-diethoxyphosphoryl-N-methylnitrone with substituted alkenes. Tetrahedron Lett. 2010, 51, 2617–2621. [Google Scholar] [CrossRef]
- Teterina, P.S.; Efremova, M.M.; Sirotkina, E.V.; Novikov, A.S.; Khoroshilova, O.V.; Molchanov, A.P. A highly efficient and stereoselective cycloaddition of nitrones to N-arylitaconimides. Tetrahedron Lett. 2019, 60, 151063. [Google Scholar] [CrossRef]
- Yahia, W.; Khorief Nacereddine, A.; Liacha, M.; Djerourou, A. A quantum-chemical DFT study of the mechanism and regioselectivity of the 1, 3-dipolar cycloaddition reaction of nitrile oxide with electron-rich ethylenes. Int. J. Quantum Chem. 2018, 118, e25540. [Google Scholar] [CrossRef]
- Khorief Nacereddine, A.; Merzoud, L.; Morell, C.; Chermette, H. A computational investigation of the selectivity and mechanism of the Lewis acid catalyzed oxa-Diels–Alder cycloaddition of substituted diene with benzaldehyde. J. Comput. Chem. 2021, 42, 1296–1311. [Google Scholar] [CrossRef]
- Lamri, S.; Heddam, A.; Kara, M.; Yahia, W.; Khorief Nacereddine, A. The Role of the Catalyst on the Reactivity and Mechanism in the Diels–Alder Cycloaddition Step of the Povarov Reaction for the Synthesis of a Biological Active Quinoline Derivative: Experimental and Theoretical Investigations. Organics 2021, 2, 57–71. [Google Scholar] [CrossRef]
- Khorief Nacereddine, A. A MEDT computational study of the mechanism, reactivity and selectivity of non-polar [3+2] cycloaddition between quinazoline-3-oxide and methyl 3-methoxyacrylate. J. Mol. Model. 2020, 6, 1–12. [Google Scholar] [CrossRef]
- Barama, L.; Bayoud, B.; Chafaa, F.; Khorief Nacereddine, A.; Djerourou, A. A mechanistic MEDT study of the competitive catalysed [4+2] and [2+2] cycloaddition reactions between 1-methyl-1-phenylallene and methyl acrylate: The role of Lewis acid on the mechanism and selectivity. Struct. Chem. 2018, 29, 1709–1721. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular electron density theory: A modern view of reactivity in organic chemistry. Molecules. 2016, 21, 1319. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, 37, 785. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Simkin, B.I.; Sheĭkhet, I.I. Quantum Chemical and Statistical Theory of Solutions—Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New dual descriptor for chemical reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Hellel, D.; Chafaa, F.; Nacereddine, A.K.; Djerourou, A.; Vrancken, E. Regio-and stereoselective synthesis of novel isoxazolidine heterocycles by 1, 3-dipolar cycloaddition between C-phenyl-N-methylnitrone and substituted alkenes. Experimental and DFT investigation of selectivity and mechanism. RSC Adv. 2017, 7, 30128–30141. [Google Scholar] [CrossRef]
- Yahia, W.; Nacereddine, A.K.; Liacha, M. Towards understanding the role of Lewis acid on the regioselectivity and mechanism for the acetylation reaction of 2-benzoxazolinone with acetyl chloride: A DFT study. Prog. React. Kinet. Mech. 2014, 39, 365–374. [Google Scholar] [CrossRef]
- Bayoud, B.; Barama, L.; Nacereddine, A.K.; Djerourou, A. Shedding light on the factors controlling the mechanism, selectivity and reactivity of the Diels–Alder reactions between substituted pyridinones and ethylenes: A MEDT study. Mol. Phys. 2021, 119, e1828635. [Google Scholar] [CrossRef]
- Sobhi, C.; Nacereddine, A.K.; Nasri, L.; Lechtar, Z.; Djerourou, A. A DFT study of the mechanism and the regioselectivity of [3+ 2] cycloaddition reactions of nitrile oxides with α, β-acetylenic aldehyde. Mol. Phys. 2016, 114, 3193–3200. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Sobhi, C.; Nacereddine, A.K.; Djerourou, A.; Aurell, M.J.; Domingo, L.R. The role of the trifluoromethyl group in reactivity and selectivity in polar cycloaddition reactions. A DFT study. Tetrahedron 2012, 68, 8457–8462. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]
- Sadi, S.; Khorief Nacereddine, A.; Djerourou, A. The effects of solvent nature and steric hindrance on the reactivity, mechanism and selectivity of the cationic imino-Diels–Alder cycloaddition reaction between cationic 2-azadienes and arylpropene. J. Phys. Org. Chem. 2022, 35, e4311. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the mechanism of polar Diels–Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P. A DFT analysis of the participation of zwitterionic TACs in polar [3+2] cycloaddition reactions. Tetrahedron 2014, 70, 4519–4525. [Google Scholar] [CrossRef]
- Benchouk, W.; Mekelleche, S.M.; Silvi, B.; Aurell, M.J.; Domingo, L.R. Understanding the kinetic solvent effects on the 1, 3-dipolar cycloaddition of benzonitrile N-oxide: A DFT study. J. Phys. Org. Chem. 2011, 24, 611–618. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactant | HOMO | LUMO | µ | η | ω | N |
|---|---|---|---|---|---|---|
| Nitrone 1 | −5.74 | −2.05 | −3.89 | 3.68 | 2.06 | 3.39 |
| Ethylene 2 | −6.39 | −1.80 | −4.09 | 4.58 | 1.83 | 2.74 |
| System | E (a.u) | ΔE (kcal/mol) | E (a.u) | ΔE (kcal/mol) |
|---|---|---|---|---|
| Gas Phase | Dichloromethane | |||
| Alkene | −629.81087 | −629.823074 | ||
| Nitrone | −879.26189 | −879.27378 | ||
| TSmn | −1509.06612 | 4.17 | −1509.08303 | 8.68 |
| TSmx | −1509.06713 | 3.54 | −1509.08269 | 8.89 |
| TSon | −1509.07438 | −1.02 | −1509.08977 | 4.45 |
| TSox | −1509.05882 | 8.75 | −1509.07558 | 13.36 |
| CAmn | −1509.11571 | −26.95 | −1509.13108 | −21.47 |
| CAmx | −1509.11574 | −26.97 | −1509.13074 | −21.26 |
| CAon | −1509.12384 | −32.05 | −1509.13821 | −25.95 |
| CAox | −1509.11167 | −24.41 | −1509.12653 | −18.62 |
| System | H | ∆H | S | ∆S | G | ∆G |
|---|---|---|---|---|---|---|
| Alkene | −629.636002 | 102.186 | −629.68453 | |||
| Nitrone | −878.976215 | 144.081 | −879.044638 | |||
| TSon | −1508.600621 | 7.28 | 209.023 | 106.837 | −1508.699885 | 18.38 |
| Tsox | −1508.560719 | 32.31 | 204.023 | 101.837 | −1508.657608 | 44.90 |
| TSmn | −1508.568092 | 27.69 | 203.132 | 100.946 | −1508.664558 | 40.54 |
| TSmx | −1508.555116 | 35.83 | 199.318 | 97.132 | −1508.649771 | 49.82 |
| Caon | −1508.645213 | −20.70 | 209.115 | 106.929 | −1508.744521 | −9.63 |
| Caox | −1508.644865 | −20.49 | 209.978 | 107.792 | −1508.744583 | −9.67 |
| Camn | −1508.652843 | −25.49 | 211.35 | 109.164 | −1508.753212 | −15.09 |
| Camx | −1508.64097 | −18.04 | 210.757 | 108.571 | −1508.741057 | −7.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khorief Nacereddine, A.; Chafaa, F. Unveiling the Origin of the Selectivity and the Molecular Mechanism in the [3+2] Cycloaddition Reaction of N-aryl-C-carbamoylnitrone with N-arylitaconimide. Organics 2022, 3, 281-292. https://doi.org/10.3390/org3030021
Khorief Nacereddine A, Chafaa F. Unveiling the Origin of the Selectivity and the Molecular Mechanism in the [3+2] Cycloaddition Reaction of N-aryl-C-carbamoylnitrone with N-arylitaconimide. Organics. 2022; 3(3):281-292. https://doi.org/10.3390/org3030021
Chicago/Turabian StyleKhorief Nacereddine, Abdelmalek, and Fouad Chafaa. 2022. "Unveiling the Origin of the Selectivity and the Molecular Mechanism in the [3+2] Cycloaddition Reaction of N-aryl-C-carbamoylnitrone with N-arylitaconimide" Organics 3, no. 3: 281-292. https://doi.org/10.3390/org3030021