
Bis(3-methylthio-1-azulenyl)phenylmethyl Cations and Dications Connected by a 1,4-Phenylene Spacer: Synthesis and Their Electrochemical Properties

Abstract
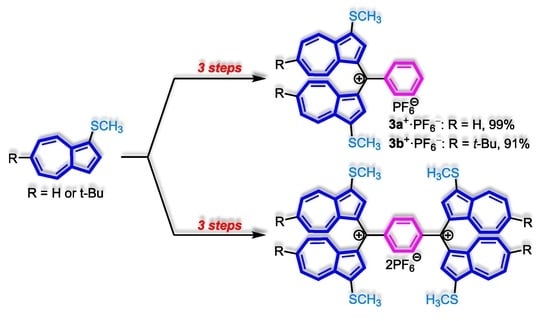
:
1. Introduction
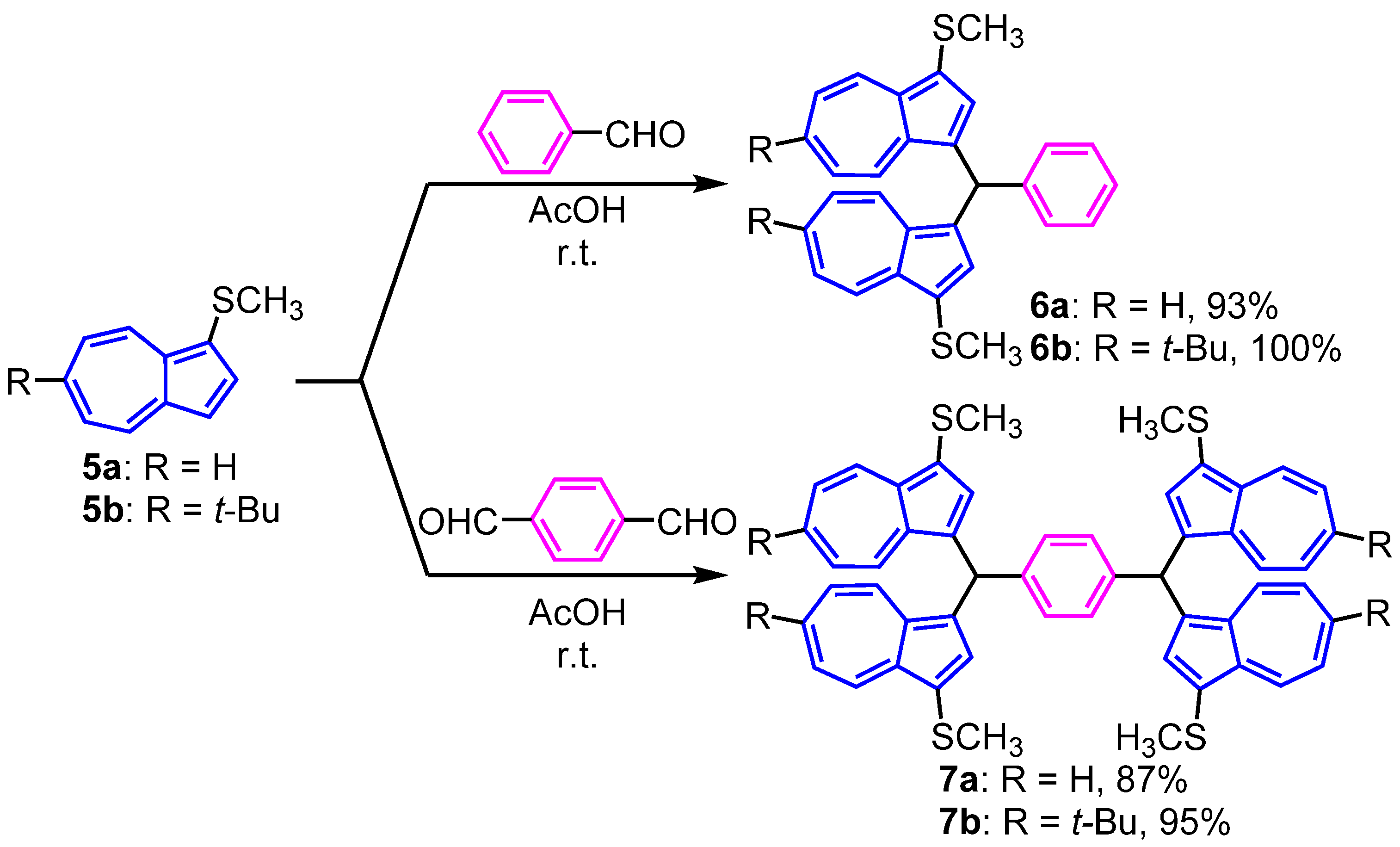
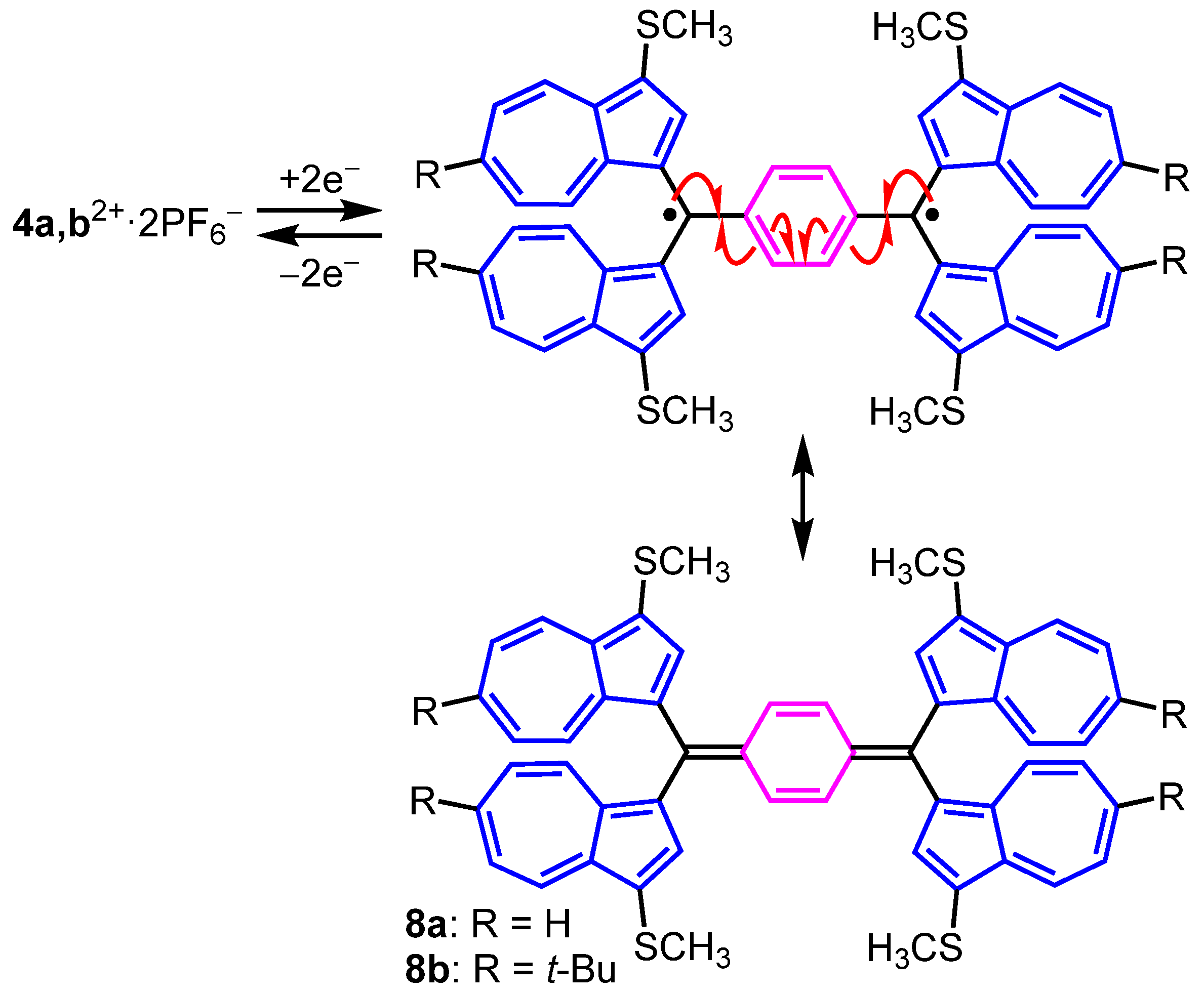
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zeller, K.-P. Azulene in Methoden. Org. Chem. 1952, V Pt 2c, 127–418. [Google Scholar]
- Ito, S.; Morita, N. Creation of Stabilized Electrochromic Materials by Taking Advantage of Azulene Skeletons. Eur. J. Org. Chem. 2009, 2009, 4567–4579. [Google Scholar] [CrossRef]
- Ito, S.; Shoji, T.; Morita, N. Recent Advances in the Development of Methods for the Preparation of Functionalized Azulenes for Electrochromic Applications. Synlett 2011, 16, 2279–2298. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S. The Preparation and Properties of Heteroarylazulenes and Hetero-Fused Azulenes. Adv. Heterocycl. Chem. 2018, 126, 1–54. [Google Scholar]
- Shoji, T.; Okujima, T.; Ito, S. Development of Heterocycle-Substituted and Fused Azulenes in the Last Decade (2010–2020). Int. J. Mol. Sci. 2020, 21, 7087. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Ito, S.; Yasunami, M. Synthesis of Azulene Derivatives from 2H-Cyclohepta[b]furan-2-ones as StartingMaterials: Their Reactivity and Properties. Int. J. Mol. Sci. 2021, 22, 10686. [Google Scholar] [CrossRef]
- Tomiyama, T.; Wakabayashi, S.; Kosakai, K.; Yokota, M. Azulene derivatives: New non-prostanoid thromboxane A2 receptor antagonists. J. Med. Chem. 1990, 33, 2323–2326. [Google Scholar] [CrossRef]
- Tomiyama, T.; Yokota, M.; Wakabayashi, S.; Kosakai, K.; Yanagisawa, T. Design, synthesis, and pharmacology of 3-substituted sodium azulene-1-sulfontes and related compounds: Non-prostanoid thromboxane A2 receptor antagonists. J. Med. Chem. 1993, 36, 791–800. [Google Scholar] [CrossRef]
- Nakamura, H.; Sekido, M.; Yamamoto, Y. Synthesis of Carboranes Containing an Azulene Framework and in vitro Evaluation as Boron Carriers. J. Med. Chem. 1997, 40, 2825–2830. [Google Scholar] [CrossRef]
- Rekka, E.; Chrysselis, M.; Siskou, I.; Kourounakis, A. Synthesis of New Azulene Derivatives and Study of Their Effect on Lipid Peroxidation and Lipoxygenase Activity. Chem. Pharm. Bull. 2002, 50, 904–907. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Kikuchi, S.; Okujima, T.; Morita, N.; Asao, T. Synthesis, Properties, and Redox Behavior of Di(1-azulenyl)(2- and 3-thienyl)methyl Cations and Dications Composed of Two Di(1-azulenyl)methylium Units Connected with 2,5-Thiophenediyl and 2,5-Thienothiophenediyl Spacers. J. Org. Chem. 2001, 66, 2470–2479. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Nomura, A.; Morita, N.; Kabuto, C.; Kobayashi, H.; Maejima, S.; Fujimori, K.; Yasunami, M. Synthesis and Two-Electron Redox Behavior of Diazuleno[2,1-a:1,2-c]naphthalenes. J. Org. Chem. 2002, 67, 7295–7302. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Inabe, H.; Morita, N.; Ohta, K.; Kitamura, T.; Imafuku, K. Synthesis of Poly(6-azulenylethynyl)benzene Derivatives as a Multielectron Redox System with Liquid Crystalline Behavior. J. Am. Chem. Soc. 2003, 125, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kubo, T.; Morita, N.; Ikoma, T.; Tero-Kubota, S.; Kawakami, J.; Tajiri, A. Azulene-Substituted Aromatic Amines. Synthesis and Amphoteric Redox Behavior of N,N-Di(6-azulenyl)-p-toluidine and N,N,N‘,N‘-Tetra(6-azulenyl)-p-phenylenediamine and Their Derivatives. J. Org. Chem. 2005, 70, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Akimoto, K.; Kawakami, J.; Tajiri, A.; Shoji, T.; Satake, H.; Morita, N. Synthesis, Stabilities, and Redox Behavior of Mono-, Di-, and Tetracations Composed of Di(1-azulenyl)methylium Units Connected to a Benzene Ring by Phenyl- and 2-Thienylacetylene Spacers. A Concept of a Cyanine−Cyanine Hybrid as a Stabilized Electrochromic System. J. Org. Chem. 2007, 72, 162–172. [Google Scholar]
- Shoji, T.; Ito, S.; Toyota, K.; Yasunami, M.; Morita, N. Synthesis, Properties, and Redox Behavior of Mono-, Bis-, and Tris[1,1,4,4,-tetracyano-2-(1-azulenyl)-3-butadienyl] Chromophores Binding with Benzene and Thiophene Cores. Chem. Eur. J. 2008, 14, 8398–8408. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S.; Toyota, K.; Iwamoto, T.; Yasunami, M.; Morita, N. Reactions between 1-Ethynylazulenes and 7,7,8,8-Tetracyanoquinodimethane (TCNQ): Preparation, Properties, and Redox Behavior of Novel Azulene-Substituted Redox-Active Chromophores. Eur. J. Org. Chem. 2009, 2009, 4316–4324. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S.; Okujima, T.; Morita, N. Synthesis of 2-Azulenyl-1,1,4,4-tetracyano-3-ferrocenyl-1,3-butadienes by [2+2] Cycloaddition of (Ferrocenylethynyl)azulenes with Tetracyanoethylene. Chem. Eur. J. 2013, 19, 5721–5730. [Google Scholar] [CrossRef] [Green Version]
- Shoji, T.; Maruyama, M.; Shimomura, E.; Maruyama, A.; Ito, S.; Okujima, T.; Toyota, K.; Morita, N. Synthesis, Properties, and Redox Behavior of Tetracyanobutadiene and Dicyanoquinodimethane Chromophores Bearing Two Azulenyl Substituents. J. Org. Chem. 2013, 78, 12513–12524. [Google Scholar] [CrossRef]
- Shoji, T.; Maruyama, M.; Maruyama, A.; Ito, S.; Okujima, T.; Toyota, K. Synthesis of 1,3-bis(tetracyano-2-azulenyl-3-butadienyl)azulenes by the [2+2] cycloaddition-retroelectrocyclization of 1,3-bis(azulenylethynyl)azulenes with tetracyanoethylene. Chem. Eur. J. 2014, 20, 11903–11912. [Google Scholar] [CrossRef] [Green Version]
- Shoji, T.; Ito, S. Azulene-Based Donor–Acceptor Systems: Synthesis, Optical, and Electrochemical Properties. Chem. Eur. J. 2017, 23, 16696–16709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Morita, N.; Asao, T. Synthesis, Properties, and Redox Behaviors of Di- and Trications Composed of Di(1-azulenyl)methylium Units Connected by p- and m-Phenylene and 1,3,5-Benzenetriyl Spacers. Bull. Chem. Soc. Jpn. 2000, 73, 1865–1874. [Google Scholar] [CrossRef]
- Shoji, T.; Higashi, J.; Ito, S.; Toyota, K.; Yasunami, M.; Fujimori, K.; Asao, T.; Morita, N. Synthesis and Redox Behavior of 1-Azulenyl Sulfides and Efficient Synthesis of 1,1’-Biazulenes. Eur. J. Org. Chem. 2008, 2008, 1242–1252. [Google Scholar] [CrossRef]
- Shoji, T.; Maruyama, A.; Maruyama, M.; Ito, S.; Okujima, T.; Higashi, J.; Toyota, K.; Morita, N. Synthesis and Properties of 6-Methoxy- and 6-Dimethylamino-1-methylthio- and 1,3-Bis(methylthio)azulenes and Triflic Anhydride-Mediated Synthesis of Their Biaryl Derivatives. Synthesis and Properties of 6-Methoxy- and 6-Dimethylamino-1-methylthio- and 1,3-Bis(methylthio)azulenes and Triflic Anhydride-Mediated Synthesis of Their Biaryl Derivatives. Bull. Chem. Soc. Jpn. 2014, 87, 141–154. [Google Scholar]
- Shoji, T.; Ito, S.; Toyota, K.; Yasunami, M.; Morita, N. The novel transition metal free synthesis of 1,1’-biazulene. Tetrahedron Lett. 2007, 48, 4999–5002. [Google Scholar] [CrossRef]
- Higashi, J.; Shoji, T.; Ito, S.; Toyota, K.; Yasunami, M.; Morita, N. Heteroarylation of 1-Azulenyl Methyl Sulfide: Two-Step Synthetic Strategy for 1-Methylthio-3-(heteroaryl)azulenes Using the Triflate of N-Containing Heterocycles. Eur. J. Org. Chem. 2008, 2008, 5823–5831. [Google Scholar] [CrossRef]
- Shoji, T.; Ito, S.; Toyota, K.; Iwamoto, T.; Yasunami, M.; Morita, N. Synthesis and Redox Behavior of 1,3-Bis(methylthio-) and 1,3-Bis(phenylthio)azulenes Bearing 2- and 3-Thienyl Substituents by Palladium-Catalyzed Cross-Coupling Reaction of 2- and 6-Haloazulenes with Thienylmagnesium Ate Complexes. Eur. J. Org. Chem. 2009, 2009, 4307–4315. [Google Scholar] [CrossRef]
- Shoji, T.; Higashi, H.; Ito, S.; Morita, N. Synthesis, Properties, and Redox Behavior of Ferrocene-Substituted Bis(3-methylthio-1-azulenyl)methylium Ions. Eur. J. Inorg. Chem. 2010, 2010, 4886–4891. [Google Scholar] [CrossRef]
- Kirby, E.C.; Reid, D.H. Conjugated cyclic hydrocarbons and their heterocyclic analogues. Part II. The condensation of azulenes with homocyclic and heterocyclic aromatic aldehydes in the presence of perchloric acid. J. Chem. Soc. 1960, 494–501. [Google Scholar] [CrossRef]
- Kirby, E.C.; Reid, D.H. Conjugated cyclic hydrocarbons and their heterocyclic analogues. Part V. 1-1′-Azulenylmethyleneazulenium salts and 1-ethoxymethyl-eneazulenium salts. J. Chem. Soc. 1961, 1724–1730. [Google Scholar] [CrossRef]
- Kirby, E.C.; Reid, D.H. Conjugated cyclic hydrocarbons and their heterocyclic analogues. Part VI. The condensation of azulenes with aliphatic aldehydes in the presence of perchloric acid. J. Chem. Soc. 1961, 3579–3593. [Google Scholar] [CrossRef]
- Takekuma, S.-i.; Takekuma, H.; Matsubara, Y.; Hirai, A.; Yamamoto, H.; Nozoe, T. Reactions of Guaiazulene with Aldehyde Reagents in Acetic Acid—An Efficient Preparation of 3,3’-Methylenediguaiazulene Derivatives. Nippon Kagaku Kaishi 1996, 1996, 419–423. [Google Scholar] [CrossRef] [Green Version]
- Takekuma, S.-i.; Takekuma, H.; Hanaoka, Y.; Yamamoto, H. Reactions of Azulene with Heteroaromatic Carbaldehydes in Acetic Acid. Nippon Kagaku Kaishi 1996, 1996, 659–662. [Google Scholar] [CrossRef] [Green Version]
- Takekuma, S.-i.; Takekuma, H.; Hatanaka, Y.; Kawaguchi, J.; Yamamoto, H. Reactions of Guaiazulene with Phthalaldehyde, Isophthalaldehyde and Terephthalaldehyde in Acetic Acid. Nippon Kagaku Kaishi 1998, 1998, 275–279. [Google Scholar] [CrossRef]
- Takekuma, S.-i.; Sasaki, M.; Takekuma, H.; Yamamoto, H. Preparation and Characteristic Properties of 1,4-Bis(3-guaiazulenylmethylium)benzene Bishexafluorophosphate. Chem. Lett. 1999, 28, 999–1000. [Google Scholar] [CrossRef]
- Ito, S.; Morita, N.; Asao, T. Azulene analogues of triphenylmethyl cation; extremely stable hydrocarbon carbocations. Tetrahedron Lett. 1991, 32, 773–776. [Google Scholar] [CrossRef]
- Cowper, P.; Pockett, A.; Kociok-Köhn, G.; Cameron, P.J.; Lewis, S.E. Azulene-Thiophene-Cyanoacrylic acid dyes with donor-π-acceptor structures. Synthesis, characterisation and evaluation in dye-sensitized solar cells. Tetrahedron 2018, 74, 2775–2786. [Google Scholar] [CrossRef]
- Suppan, P.; Ghoneim, N. Solvatochromism, The Royal Society of Chemistry; Londres: Cambridge, UK, 1997. [Google Scholar]
- Suppan, P. Invited review solvatochromic shifts: The influence of the medium on the energy of electronic states. J. Photochem. Photobiol. A 1990, 50, 293–330. [Google Scholar] [CrossRef]
- Christian, R. Solvent and Solvent Effects in Organic Chemistry; Wiley-VCH: New York, NY, USA, 2004. [Google Scholar]
- Takekuma, S.-I.; Sasaki, M.; Takekuma, H.; Yamamoto, H. The isolation of quinoid compound was accomplished by Takekuma. An Efficient Preparation and Characteristic Properties of Tetramethyl 3,3′,3″,3‴-(p–Quinodimethane-7,7,8,8-tetrayltetraazulene-1,1′,1″,1‴-tetracarboxylate. Nippon Kagaku Kaishi 2000, 2000, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, K.; Ohta, K.; Fujimoto, T.; Yamamoto, I. Chromic materials. Part 1.-Liquid-crystalline behaviour and electrochromism in bis(octakis-n-alkylphthalocyaninato)lutetium(III) complexes. J. Mater. Chem. 1994, 4, 533–536. [Google Scholar] [CrossRef]
- Rosseinsky, D.R.; Monk, P.M.S. Studies of tetra-(bipyridilium) salts as possible polyelectrochromic materials. J. Appl. Electrochem. 1994, 24, 1213–1221. [Google Scholar] [CrossRef]
- Monk, P.M.S.; Mortimer, R.J.; Rosseinsky, D.R. Electrochromism and Electrochromic Devices; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Hünig, S.; Kemmer, M.; Wenner, H.; Perepichka, I.F.; Bäuerle, P.; Emge, A.; Gescheidt, G. Violene/Cyanine Hybrids: A General Structure for Electrochromic Systems. Chem. Eur. J. 1999, 5, 1969–1973. [Google Scholar] [CrossRef]
- Hünig, S.; Kemmer, M.; Wenner, H.; Barbosa, F.; Gescheidt, G.; Perepichka, I.F.; Bäuerle, P.; Emge, A.; Peters, K. Violene/Cyanine Hybrids as Electrochromics Part 2: Tetrakis(4-dimethylaminophenyl)ethene and Its Derivatives. Chem. Eur. J. 2000, 6, 2618–2632. [Google Scholar] [CrossRef] [PubMed]
- Hunig, S.; Perepichka, I.F.; Kemmer, M.; Wenner, H.; Bauerle, P.; Emge, A. Violene/cyanine hybrids as electrochromic systems. Part 3: Heterocyclic onium end groups. Tetrahedron 2000, 56, 4203–4211. [Google Scholar] [CrossRef]
- Hünig, S.; Briehn, C.A.; Bäuerle, P.; Emge, A. Electrochromics by Intramolecular Redox Switching of Single Bonds. Chem. Eur. J. 2001, 7, 2745–2757. [Google Scholar] [CrossRef]
- Hünig, S.; Langels, A.; Schmittel, M.; Wenner, H.; Perepichka, I.F.; Peters, K. Violene/Cyanine Hybrids as Electrochromic Systems: A New Variation of the General Structure. Eur. J. Org. Chem. 2001, 2001, 1393–1399. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
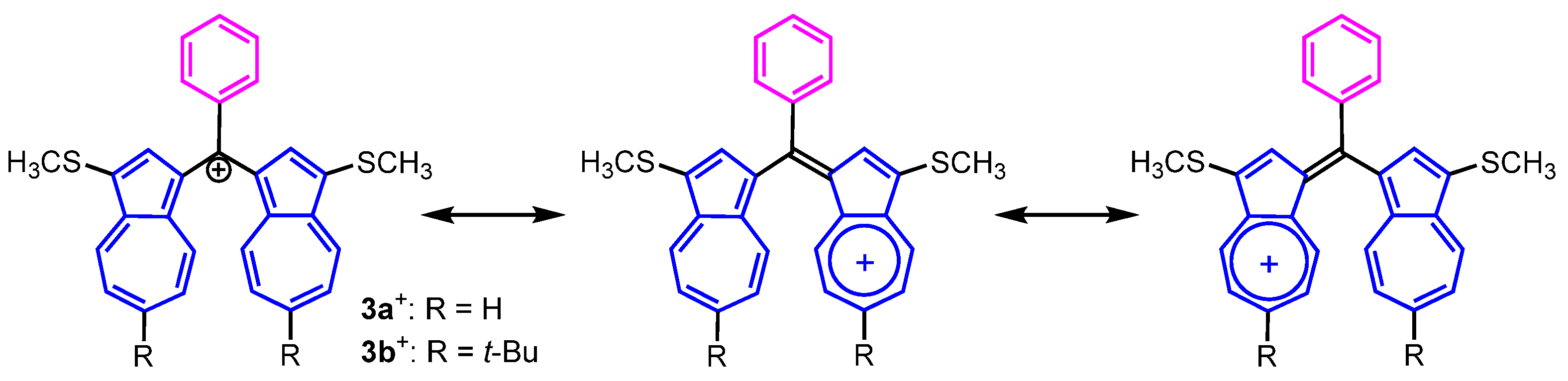{kind=link}
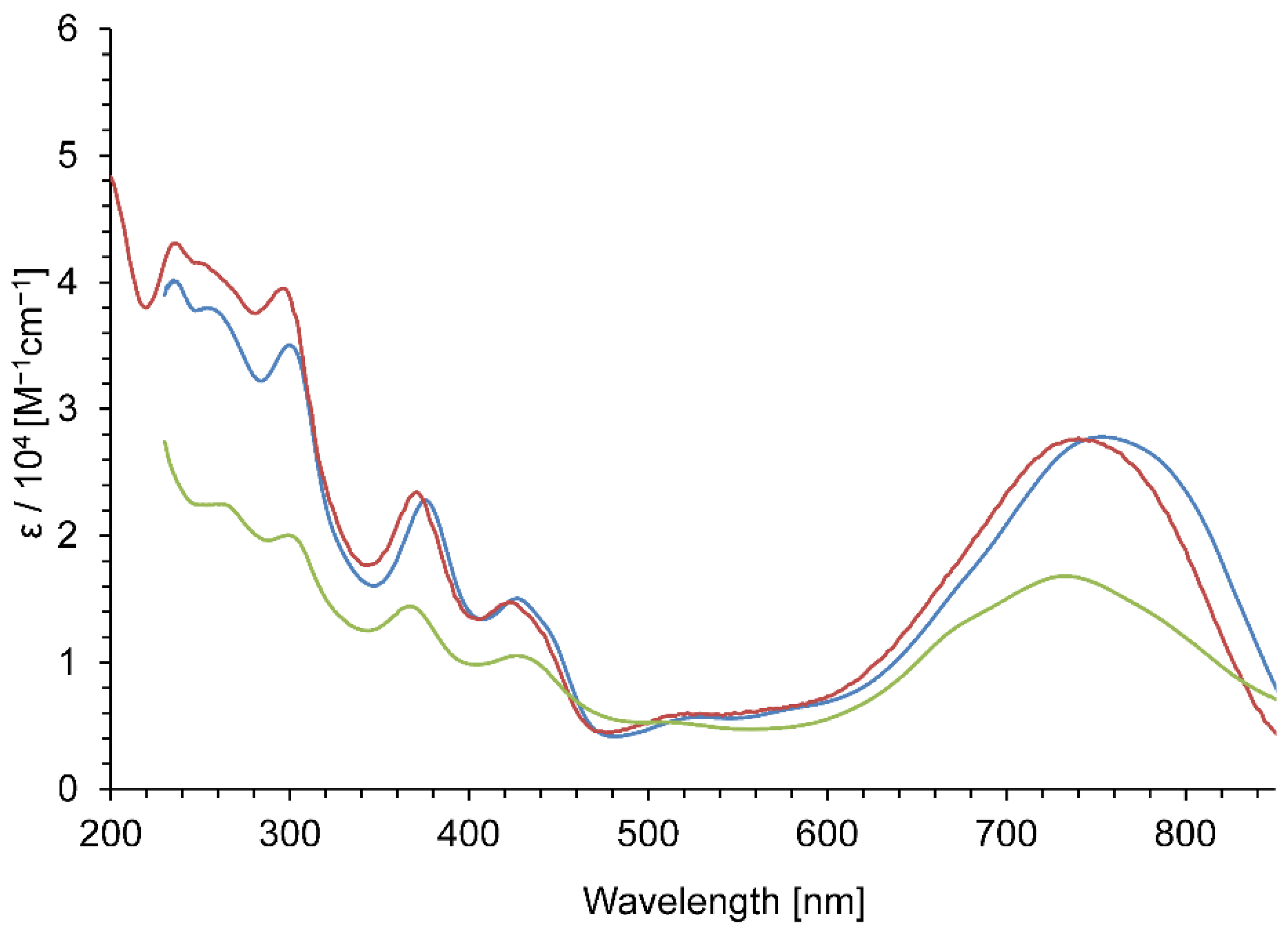{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| λmax, nm (log ε) | |||
|---|---|---|---|
| Compound | CH2Cl2 | MeCN | Hexane a |
| 3a+·PF6− | 755 (4.44) | 740 (4.44) | 732 (4.42) |
| 3b+·PF6− | 740 (4.40) | 738 (4.44) | 723 (4.38) |
| 4a2+·2PF6− | 785 (4.55) | 773 (4.55) | − b |
| 4b2+·2PF6− | 777 (4.54) | 766 (4.62) | 766 (4.44) |
| 1+·PF6− [22] | − | 681 (4.61) | − |
| 22+·2PF6− [22] | − | 703 (4.85) | − |
| Sample | pKR+ | Sample | pKR+ |
|---|---|---|---|
| 3a+·PF6− | 9.9 ± 0.1 | 4a2+·2PF6− | 9.8 ± 0.1 |
| 3b+·PF6− | 11.6 ± 0.1 | 4b2+·2PF6− | 11.7 ± 0.1 |
| 1+·PF6− [22] | 12.4 | 22+·2PF6− [22] | 12.1 ± 0.2 |
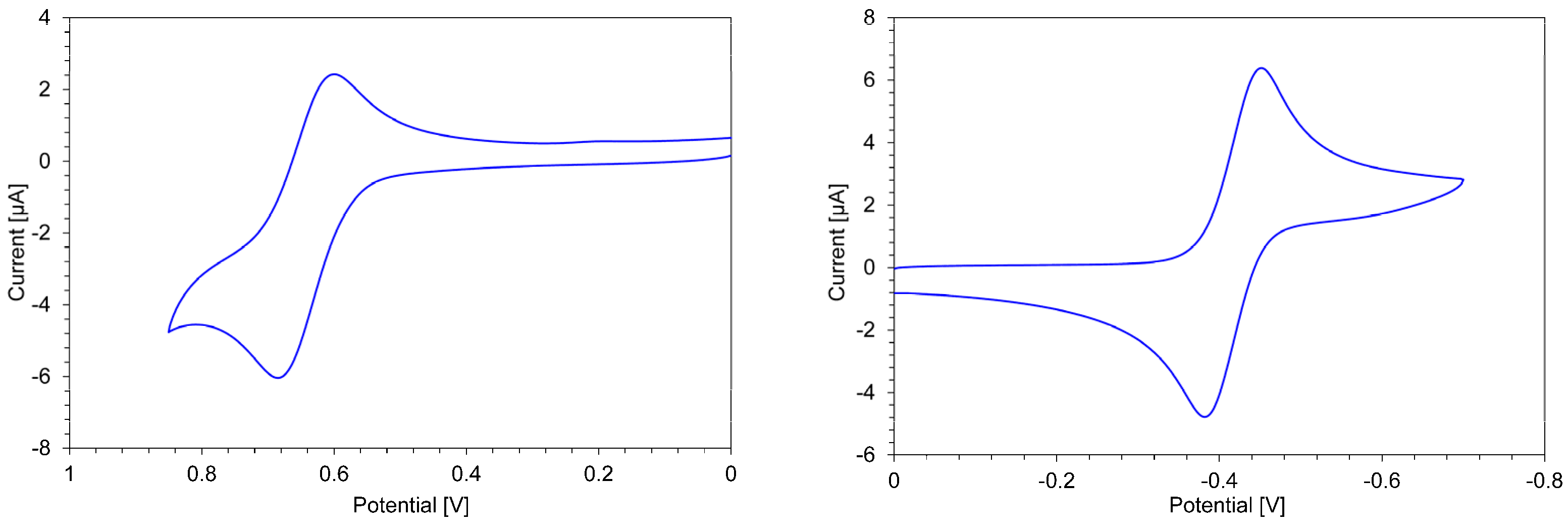
| Sample | Method | E1red [V] | E2red [V] | E1ox [V] | E2ox [V] |
|---|---|---|---|---|---|
| 3a+·PF6− | CV | –0.60 | |||
| (DPV) | (–0.59) | (–1.71) | (+0.65) | (+0.96) | |
| 3b+·PF6− | CV | –0.68 | |||
| (DPV) | (–0.66) | (–1.45) | (+0.64) | (+0.97) | |
| 4a2+·2PF6− | CV | –0.35 | |||
| (DPV) | (–0.33) | (–1.65) | (+0.67) | ||
| 4b2+·2PF6− | CV | –0.42 | +0.65 | ||
| (DPV) | (–0.40) | (–1.74) | (+0.63) | ||
| 1+·PF6− [23] b | CV b | –0.78 | +0.88 | ||
| 22+·2PF6− [23] b | CV b | –0.55 | +0.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoji, T.; Sakata, N.; Sekiguchi, R.; Ito, S. Bis(3-methylthio-1-azulenyl)phenylmethyl Cations and Dications Connected by a 1,4-Phenylene Spacer: Synthesis and Their Electrochemical Properties. Organics 2022, 3, 507-519. https://doi.org/10.3390/org3040034
Shoji T, Sakata N, Sekiguchi R, Ito S. Bis(3-methylthio-1-azulenyl)phenylmethyl Cations and Dications Connected by a 1,4-Phenylene Spacer: Synthesis and Their Electrochemical Properties. Organics. 2022; 3(4):507-519. https://doi.org/10.3390/org3040034
Chicago/Turabian StyleShoji, Taku, Naoko Sakata, Ryuta Sekiguchi, and Shunji Ito. 2022. "Bis(3-methylthio-1-azulenyl)phenylmethyl Cations and Dications Connected by a 1,4-Phenylene Spacer: Synthesis and Their Electrochemical Properties" Organics 3, no. 4: 507-519. https://doi.org/10.3390/org3040034