
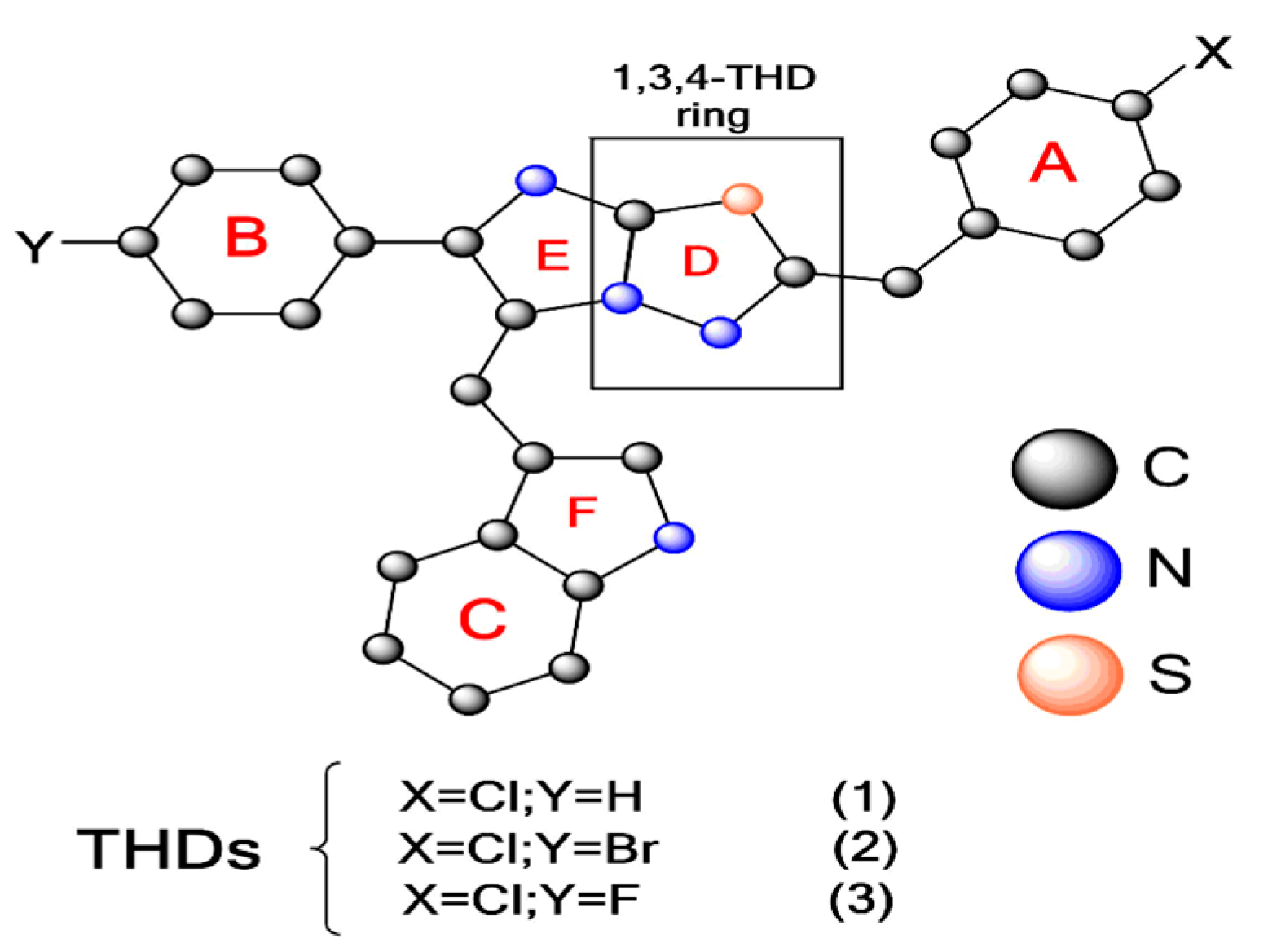
The Thiadiazole Ring (THD) Is a Building Block for Potential Inhibitors of the SARS-CoV-2 Main Protease (Mpro): Theoretical Look into the Structure, Reactivity, and Binding Profile of Three 1,3,4-THD Derivatives toward Mpro †

 ,
,
Abstract
:1. Introduction
2. Methodology
2.1. Density Functional Theory (DFT) Calculations
2.2. Molecular Docking
2.3. Molecular Dynamics Simulation
3. Results and Discussion
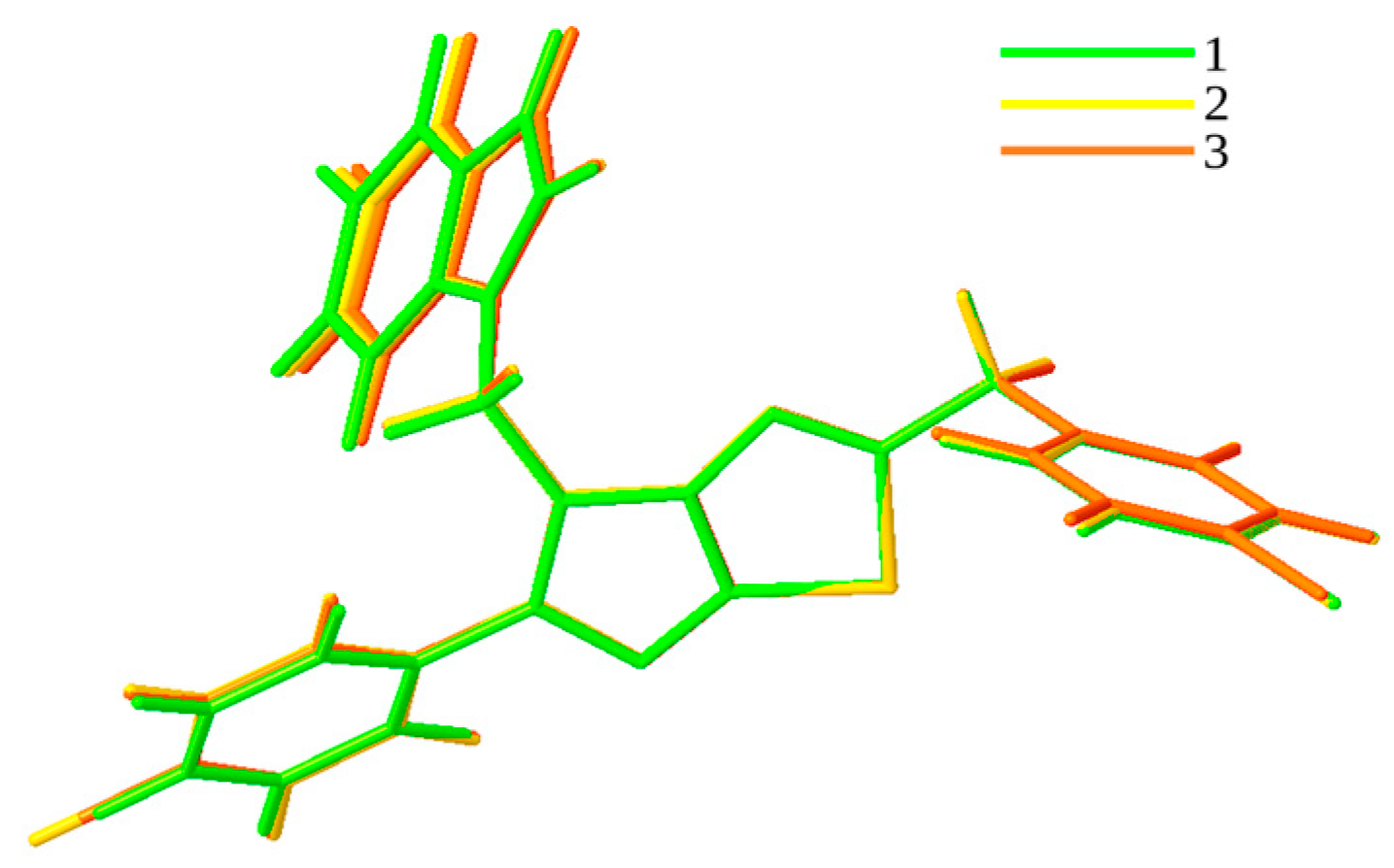
3.1. QTAIM and RDG Analyses for Dimers
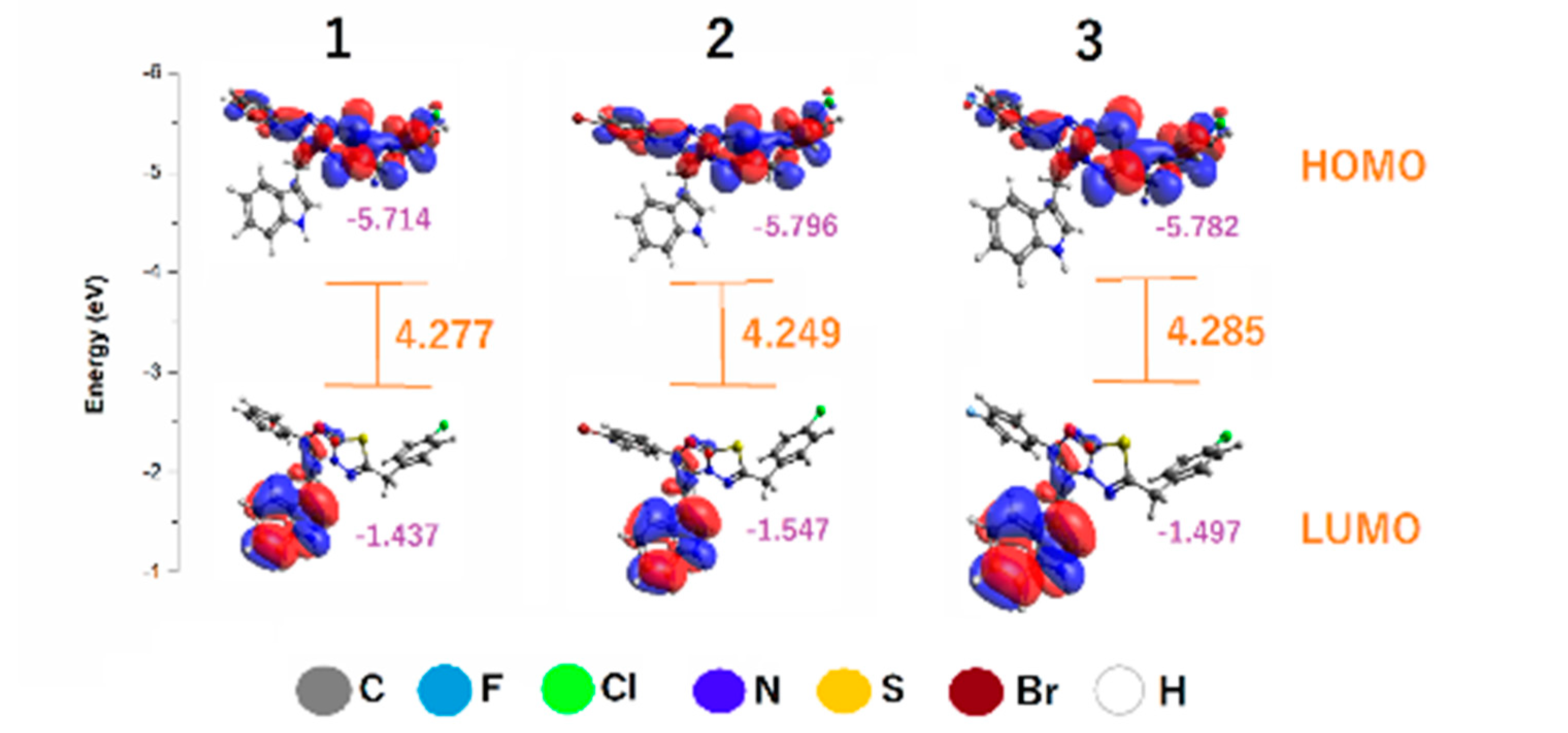
3.2. Chemical Reactivity
- (a)
- Frontier molecular orbitals
- (b)
- Molecular electrostatic potential (MEP)
- (c)
- Globalreactivity
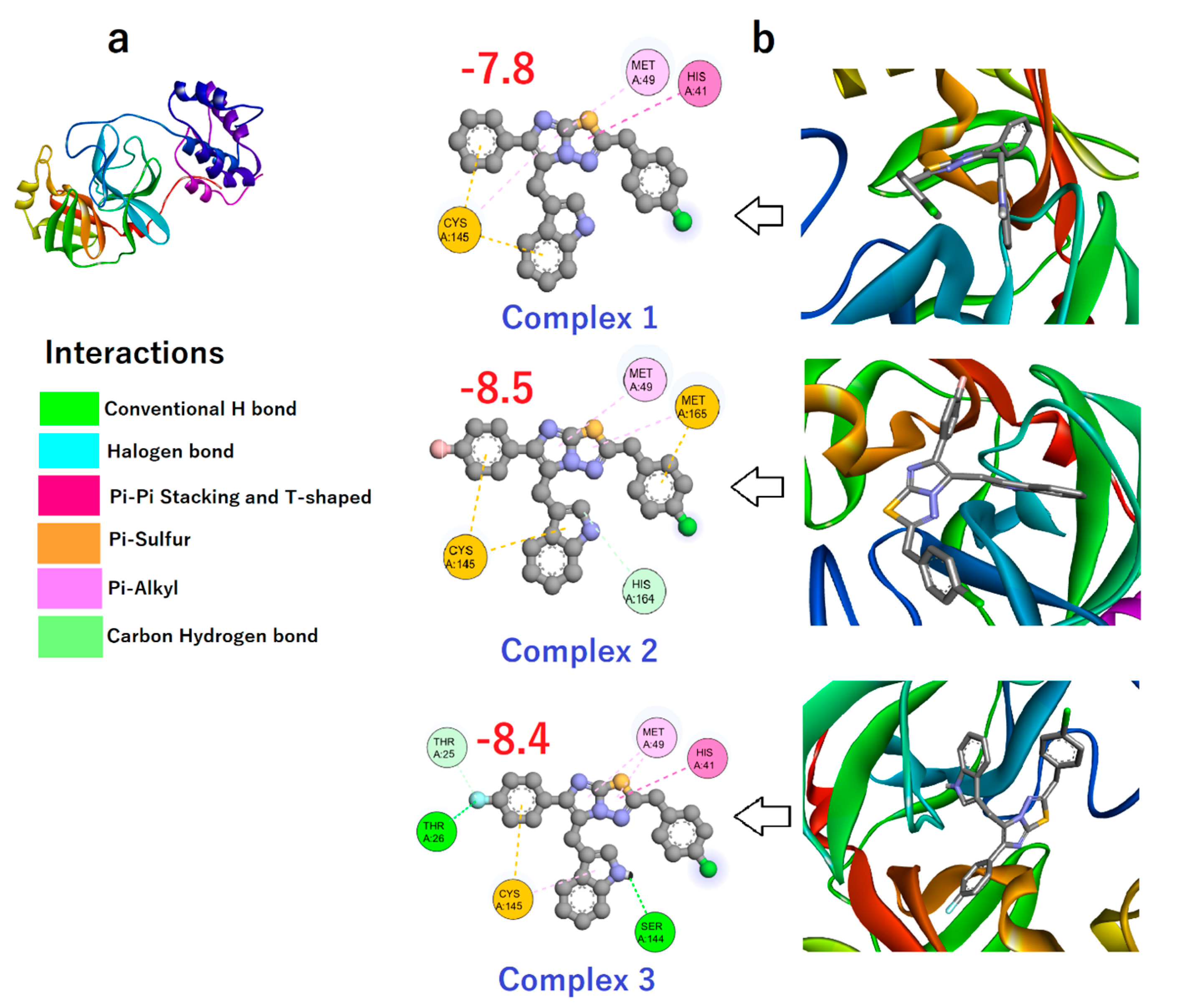
3.3. Binding Affinity of Compounds 1–3 toward Mpro
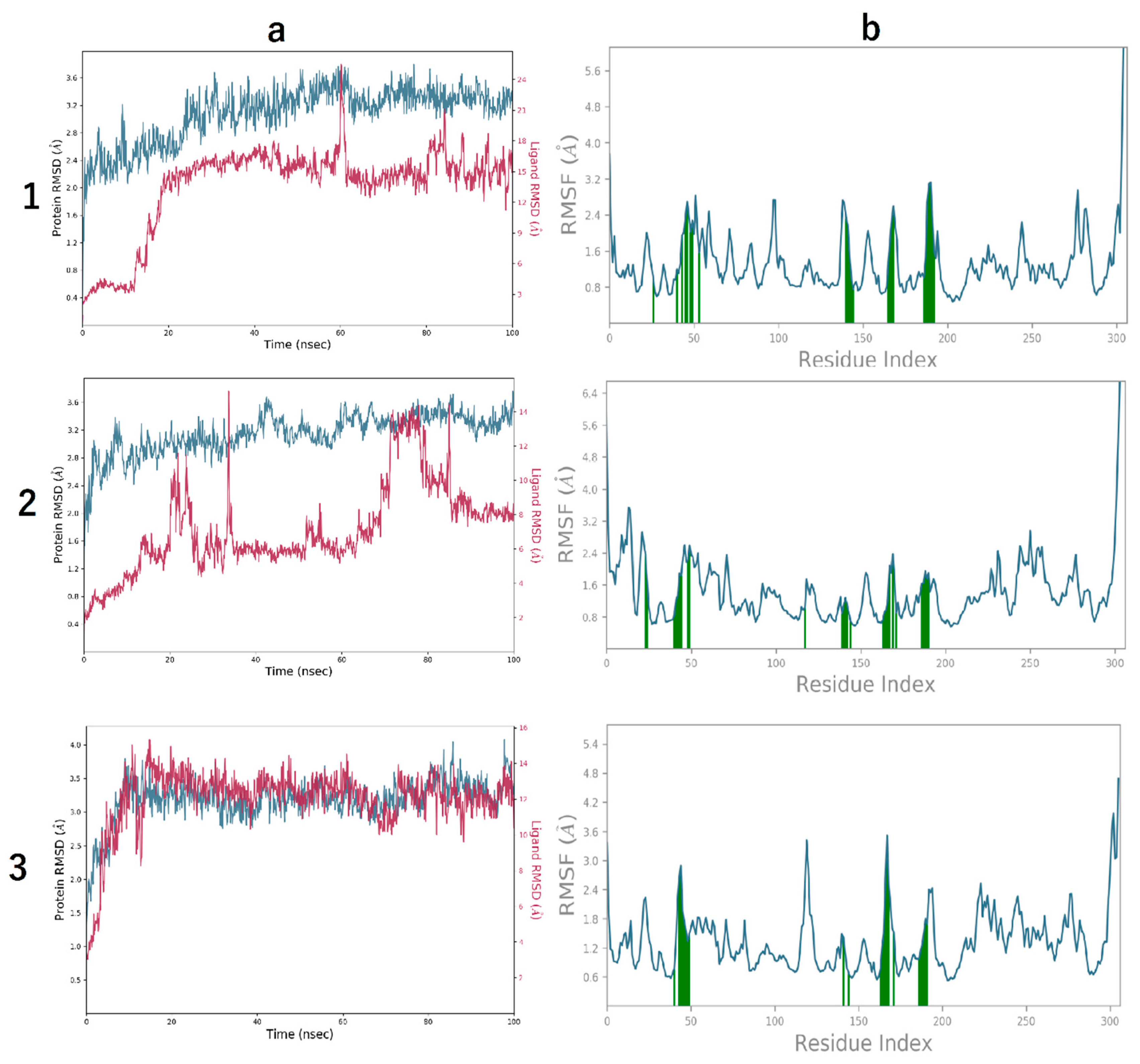
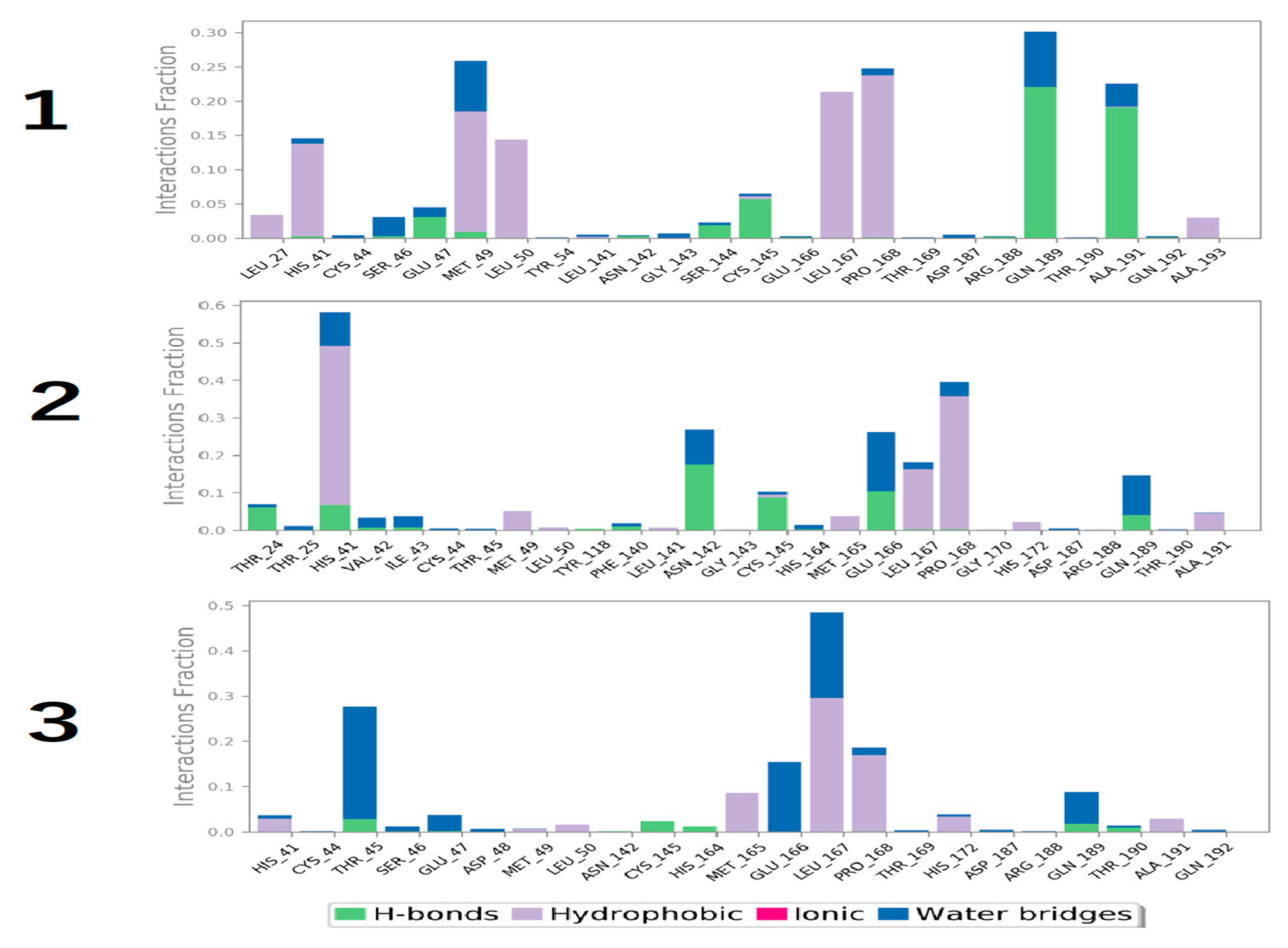
3.4. Molecular Dynamics Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fouda, A.S.; Heakal, F.E.; Radwan, M.S. Role of some thiadiazole derivatives as inhibitors for the corrosion of C-steel in 1 M H2SO4. J. Appl. Electrochem. 2009, 39, 391–402. [Google Scholar] [CrossRef]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole-a Promising Structure in Medicinal Chemistry. ChemMedChem 2013, 8, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Kumar, H.; Javed, S.A. Non-carboxylic Analogues of Naproxen: Design,Synthesis and Pharmacological Evaluation of some 1,3,4-oxadiazole/thiadiazole and 1,2,4-thiadiazole derivatives. Arch. Pharm. Chem. Life Sci. 2007, 340, 557–585. [Google Scholar] [CrossRef] [PubMed]
- Szeliga, M. Thiadiazole derivatives as anticancer agents. Pharmacol. Rep. 2020, 72, 1079–1100. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Tripathi, R.; Pandey, D.; Mishra, P. Synthesis, characterization, antimicrobial, and pharmacological evaluation of some 2, 5-disubstituted sulfonyl amino 1,3,4-oxadiazole and 2-amino-disubstituted 1,3,4-thiadiazole derivatives. J. Adv. Pharm. Technol. Res. 2014, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Shawali, A.S. 1,3,4-Thiadiazoles of pharmacological interest: Recent trends in their synthesis via tandem 1,3-dipolar cycloaddition: Review. J. Adv. Res. 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Brullo, C.; Bruno, O.; Bondavalli, F.; Ranise, A.; Filippelli, W.; Rinaldi, B.; Capuano, A.; Falcone, G. New 1,3,4-thiadiazole derivatives endowed with analgesic and anti-inflammatory activities. Bioorganic. Med. Chem. 2006, 14, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Kolavi, G.; Hegde, V.; Khazi, I.A.; Gadad, P. Synthesis and evaluation of antitubercular activity of imidazo[2,1-b,1,3, 4]thiadiazole derivatives. Bioorganic. Med. Chem. 2006, 14, 3069–3080. [Google Scholar] [CrossRef]
- Sun, J.; Yang, Y.S.; Li, W.; Zhang, Y.B.; Wang, X.L.; Tang, J.F.; Zhu, H.L. Synthesis, biological evaluation and molecular docking studies of 1,3,4-thiadiazole derivatives containing 1,4-benzodioxan as potential antitumor agents. Bioorganic Med. Chem. Lett. 2011, 21, 6116–6121. [Google Scholar] [CrossRef]
- Grimme, S.; Schreiner, P.R. Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chemie—Int. Ed. 2018, 57, 4170–4176. [Google Scholar] [CrossRef]
- Abdel-Hamid, M.K.; Abdel-Hafez, A.A.; El-Koussi, N.A.; Mahfouz, N.M.; Innocenti, A.; Supuran, C.T. Design, synthesis, and docking studies of new 1,3,4-thiadiazole-2-thione derivatives with carbonic anhydrase inhibitory activity. Bioorganic Med. Chem. 2007, 15, 6975–6984. [Google Scholar] [CrossRef] [PubMed]
- Rastija, V.; Medić-Šarić, M. QSAR study of antioxidant activity of wine polyphenols. Eur. J. Med. Chem. 2009, 44, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Shamanth, S.; Mantelingu, K.; Kumar, H.K.; Yathirajan, H.S.; Foro, S.; Glidewell, C. Crystal structures of three 6-aryl-2-(4-chlorobenzyl)-5-[(1H-indol-3-yl)methyl]imidazo[2,1-b,1,3,4]thiadiazoles Shamanth Sadashivamurthy. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Nkungli, N.K.; Ghogomu, J.N. Theoretical analysis of the binding of iron(III) protoporphyrin IX to 4-methoxyacetophenone thiosemicarbazone via DFT-D3, MEP, QTAIM, NCI, ELF, and LOL studies. J. Mol. Model. 2017, 23, 200. [Google Scholar] [CrossRef] [PubMed]
- Saleh, G.; Gatti, C.; Presti, L.L.; Garcia, J.C. Revealing Non-covalent Interactions in Molecular Crystals through Their Experimental Electron Densities. Chem.—Eur. J. 2012, 18, 15523–15536. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 (C.01) Inc., Wallingford, CT, USA. 2009. Available online: https://gaussian.com/ (accessed on 1 December 2023).
- Wazzan, N.A.; Obot, I.B.; Kaya, S. Theoretical modeling and molecular level insights into the corrosion inhibition activity of 2-amino-1,3,4-thiadiazole and its 5-alkyl derivatives. J. Mol. Liq. 2016, 221, 579–602. [Google Scholar] [CrossRef]
- Dennington, J.M.M.R.; Keith, T.A. GaussView, version 6.0. 16; Semichem Inc.: Shawnee Mission, KS, USA, 2016.
- Hanwell, M.D.; Curtis, D.E.; Loni, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sandeep, G.; Nagasree, K.P.; Hanisha, M.; Kumar, M.M.K. AUDocker LE: A GUI for virtual screening with AUTODOCK Vina. BMC Res. Notes 2011, 4, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Castro-Alvarez, A.; Costa, A.M.; Vilarrasa, J. The Performance of several docking programs at reproducing protein-macrolide-like crystal structures. Molecules 2017, 22, 136. [Google Scholar] [CrossRef]
- Matondo, A.; Dendera, W.; Isamura, B.K.; Ngbolua, K.; Mambo, H.V.S.; Muzomwe, M.; Mudogo, V. In silico Drug Repurposing of Anticancer Drug 5-FU and Analogues Against SARS-CoV-2 Main Protease: Molecular Docking, Molecular Dynamics Simulation, Pharmacokinetics and Chemical Reactivity Studies. Adv. Appl. Bioinform. Chem. 2022, 15, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, M.; Alfaro, I.; Angel, C. Identification of potential inhibitors of SARS-CoV-2 papain-like protease from tropane alkaloids from Schizanthus porrigens: A molecular docking study. Chem. Phys. Lett. 2020, 761, 138068. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Harder, E.; Damm, W.; Friesner, R.A.; Sherman, W. Improving the prediction of absolute solvation free energies using the next generation opls force field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Czeleń, P. Inhibition mechanism of CDK-2 and GSK-3β by a sulfamoylphenyl derivative of indoline—A molecular dynamics study. J. Mol. Model. 2017, 23, 230. [Google Scholar] [CrossRef] [PubMed]
- Lohith, T.N.; Hema, M.K.; Karthik, C.S.; Sandeep, S.; Mallesha, L.; Alsaiari, N.S.; Sridhar, M.A.; Katubi, K.M.; Abualnaja, K.M.; Lokanath, N.K.; et al. Persistent prevalence of non-covalent interaction in pyrimidine containing sulfonamide derivative: A quantum computational analysis. J. Mol. Struct. 2022, 1266, 133378. [Google Scholar] [CrossRef]
- Contreras-García, J.; Yang, W.; Johnson, E.R. Analysis of hydrogen-bond interaction potentials from the electron density: Integration of noncovalent interaction regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef]
- Kasende, O.E.; Matondo, A.; Muzomwe, M.; Muya, J.T.; Scheiner, S. Interaction between temozolomide and water: Preferred binding sites. Comput. Theor. Chem. 2014, 1034, 26–31. [Google Scholar] [CrossRef]
- Geerlings, P.; Fias, S.; Boisdenghien, Z.; De Proft, F. Conceptual DFT: Chemistry from the linear response function. Chem. Soc. Rev. 2014, 43, 4989–5008. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F. Conceptual DFT: The chemical relevance of higher response functions. Phys. Chem. Chem. Phys. 2008, 10, 3028–3042. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Rong, C.; Lu, T. Information conservation principle determines electrophilicity, nucleophilicity, and regioselectivity. J. Phys. Chem. A 2014, 118, 3698–3704. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Rong, C.; Chattaraj, P.K.; Liu, S. A comparative study to predict regioselectivity, electrophilicity and nucleophilicity with Fukui function and Hirshfeld charge. Theor. Chem. Acc. 2019, 138, 124. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Quantitative Analyses of Molecular Surface Electrostatic Potentials in Relation to Hydrogen Bonding and Co-Crystallization. Cryst. Growth Des. 2015, 15, 3767–3774. [Google Scholar] [CrossRef]
- Isamura, B.K.; Lobb, K.A.; Muya, J.T. Regioselectivity, chemical bonding and physical nature of the interaction between imidazole and XAHs (X=H, F, Cl, Br, CH3, and A=S, Se, Te). Mol. Phys. 2022, 120, e2026511. [Google Scholar] [CrossRef]
- Isamura, B.K.; Patouossa, I.; Muya, J.T.; Lobb, K.A. Unveiling the reactivity of truxillic and truxinic acids (TXAs): Deprotonation, anion…H–O, cation…O and cation… π interactions in TXA0…Y+ and TXA0…Z− complexes (Y = Li, Na, K; Z = F, Cl, Br). Struct. Chem. 2022, 34, 97–112. [Google Scholar] [CrossRef]
- Kumar, S.; Choudhary, M. Synthesis and characterization of novel copper(ii) complexes as potential drug candidates against SARS-CoV-2 main protease. New J. Chem. 2022, 46, 4911–4926. [Google Scholar] [CrossRef]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE 2020, 15, e0240653. [Google Scholar] [CrossRef]
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.E.; Tawfeek, N.; Elbaramawi, S.S.; Fikry, E. Agathis robusta Bark Essential Oil Effectiveness Against COVID-19: Chemical Composition, In Silico and In Vitro Approaches. Plants 2022, 11, 663. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BCP | ρBCP (a.u.) | ∇2ρ (a.u) | V(r) (a.u.) | G(r) (a.u.) | λ1 | λ2 | λ3 | EHB (kcal/mL) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 113 | 0.0023 | 0.0065 | −0.0009 | 0.00128 | 1.422 | −0.0012 | −0.0004 | 0.0078 | 0.153 | −0.229 |
| 136 | 0.0076 | 0.0362 | −0.0040 | 0.00656 | 1.64 | −0.0047 | −0.0035 | 0.0445 | 1.056 | −0.953 | |
| 189 | 0.0207 | 0.0669 | −0.0150 | 0.0158 | 1.053 | −0.025 | −0.0233 | 0.115 | 0.217 | −3.875 | |
| 191 | 0.0057 | 0.0193 | −0.0025 | 0.0036 | 1.44 | −0.0034 | −0.0032 | 0.026 | 0.130 | −0.529 | |
| 197 | 0.0005 | 0.0017 | −0.00018 | 0.0003 | 1.666 | −0.0002 | −0.0001 | 0.00216 | 0.009 | −0.630 | |
| 2 | 152 | 0.00220 | 0.0067 | −0.00089 | 0.00128 | 1.438 | −0.0012 | −0.0008 | 0.00882 | 0.136 | −0.251 |
| 137 | 0.00383 | 0.0100 | −0.0017 | 0.00214 | 1.258 | −0.0018 | −0.0004 | 0.0123 | 0.146 | −0.112 | |
| 189 | 0.0062 | 0.0222 | −0.00355 | 0.00455 | 1.281 | −0.0048 | −0.0017 | 0.0288 | 0.166 | −0.640 | |
| 207 | 0.00319 | 0.0093 | −0.00128 | 0.00181 | 1.414 | −0.0018 | −0.0012 | 0.0125 | 0.144 | −0.003 | |
| 175 | 0.0086 | 0.0373 | −0.0045 | −0.00692 | 1.537 | −0.0072 | −0.0052 | 0.0497 | 0.144 | −1.176 | |
| 3 | 150 | 0.0079 | 0.0382 | −0.0043 | 0.0069 | 1.604 | −0.0042 | −0.0028 | 0.0453 | 0.09 | −1.02 |
| 224 | −0.0040 | 0.0100 | −0.0016 | 0.0020 | 1.25 | −0.0018 | −0.0009 | 0.0127 | 0.142 | −0.150 | |
| 178 | 0.0068 | −0.0227 | −0.0036 | 0.0046 | 1.277 | −0.0062 | −0.0056 | 0.0346 | 0.179 | −0.774 | |
| 131 | 0.0040 | 0.0099 | −0.00168 | 0.0020 | 1.190 | −0.0018 | −0.0010 | 0.0127 | 0.141 | −0.150 |
| Compounds | IP | EA | S | |||
|---|---|---|---|---|---|---|
| 1 | 5.714 | 1.438 | 2.138 | 0.468 | −3.576 | 2.990 |
| 2 | 5.796 | 1.547 | 2.125 | 0.471 | −3.672 | 3.172 |
| 3 | 5.782 | 1.497 | 2.143 | 0.467 | −3.639 | 3.091 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shanthakumar, D.C.; Nagarajappa, L.T.; Isamura, B.K.; Leoma, M.B.; Mokgopa, K.P.; Anandalwar, S.M.; Doreswamy, S.; Ningaiah, S. The Thiadiazole Ring (THD) Is a Building Block for Potential Inhibitors of the SARS-CoV-2 Main Protease (Mpro): Theoretical Look into the Structure, Reactivity, and Binding Profile of Three 1,3,4-THD Derivatives toward Mpro. Eng. Proc. 2023, 59, 94. https://doi.org/10.3390/engproc2023059094
Shanthakumar DC, Nagarajappa LT, Isamura BK, Leoma MB, Mokgopa KP, Anandalwar SM, Doreswamy S, Ningaiah S. The Thiadiazole Ring (THD) Is a Building Block for Potential Inhibitors of the SARS-CoV-2 Main Protease (Mpro): Theoretical Look into the Structure, Reactivity, and Binding Profile of Three 1,3,4-THD Derivatives toward Mpro. Engineering Proceedings. 2023; 59(1):94. https://doi.org/10.3390/engproc2023059094
Chicago/Turabian StyleShanthakumar, Dileep Chikkur, Lohith Tumakuru Nagarajappa, Bienfait Kabuyaya Isamura, Mofeli Benedict Leoma, Kabelo Phuti Mokgopa, Sridhar Mandayam Anandalwar, Sahana Doreswamy, and Srikantamurthy Ningaiah. 2023. "The Thiadiazole Ring (THD) Is a Building Block for Potential Inhibitors of the SARS-CoV-2 Main Protease (Mpro): Theoretical Look into the Structure, Reactivity, and Binding Profile of Three 1,3,4-THD Derivatives toward Mpro" Engineering Proceedings 59, no. 1: 94. https://doi.org/10.3390/engproc2023059094