

Anticancer Effects of Fucoxanthin in a PDX Model of Advanced Stage Pancreatic Cancer with Alteration of Several Multifunctional Molecules

 ,
,  ,
,  , ,
, ,  ,
,
Abstract
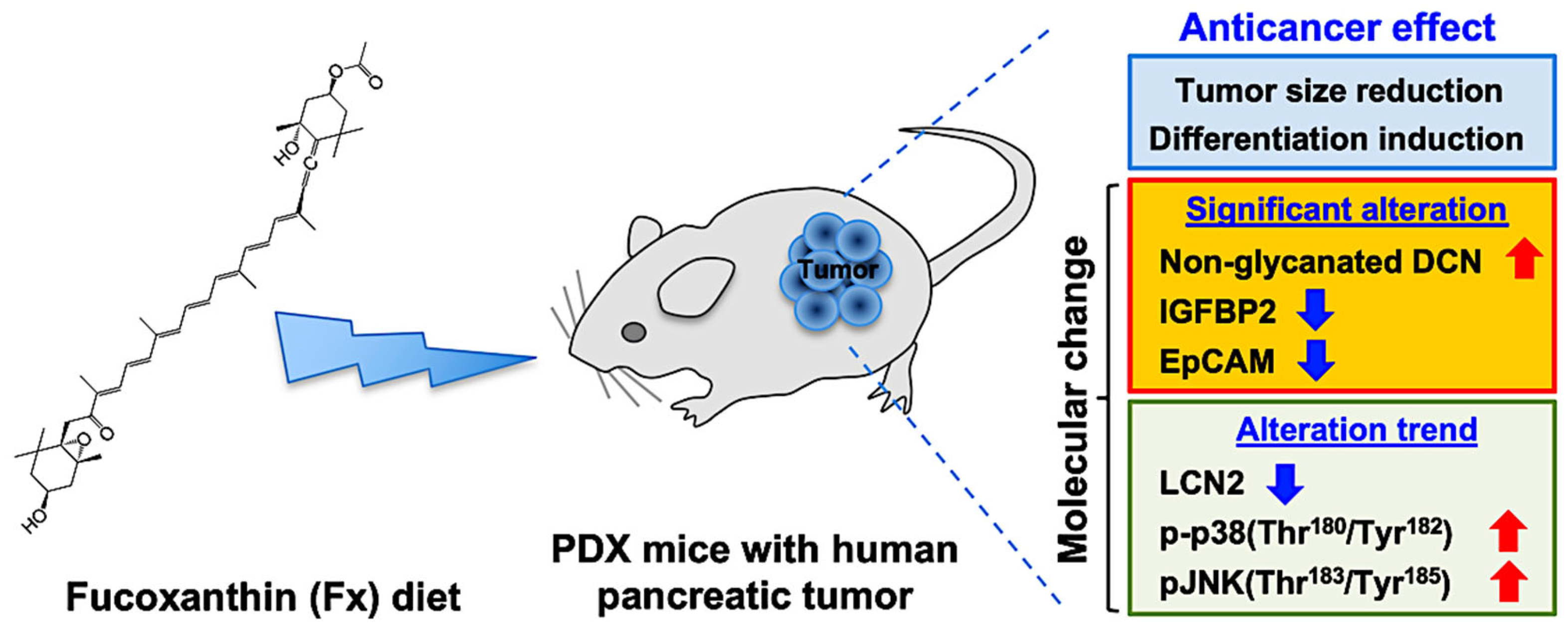
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemical
2.2. Establishment of PDX Tumor from Human PC Tissue
2.3. Animal Experiments
2.4. Proteome Analysis
2.5. Cell Proliferation Assay
2.6. Western Blot Analysis
2.7. Statistics Analysis
3. Results
3.1. Characteristics of a Tumor Derived from a Patient with PC
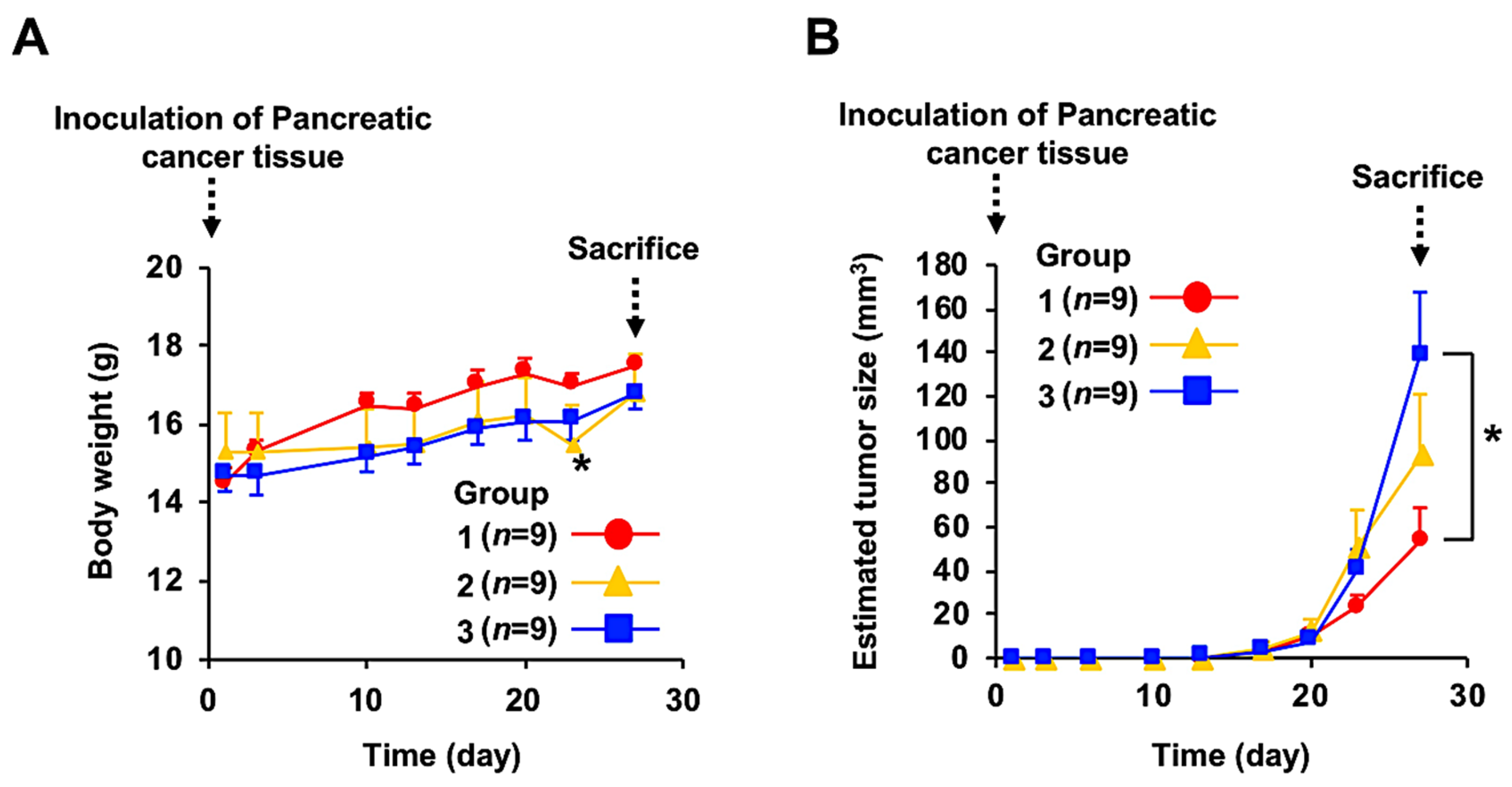
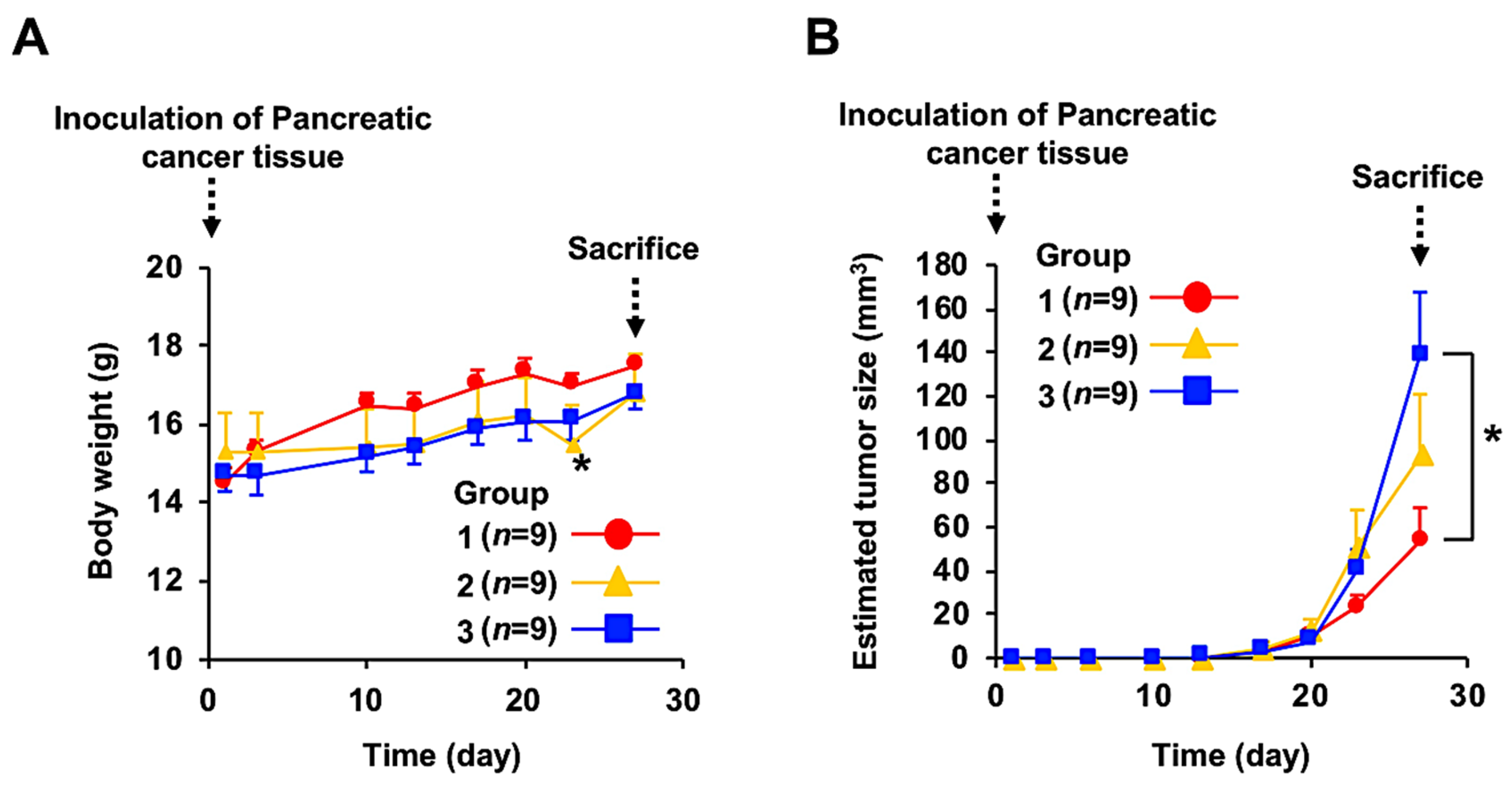
3.2. Anticancer Effect of Fx Administration on the PC-PDX Mice
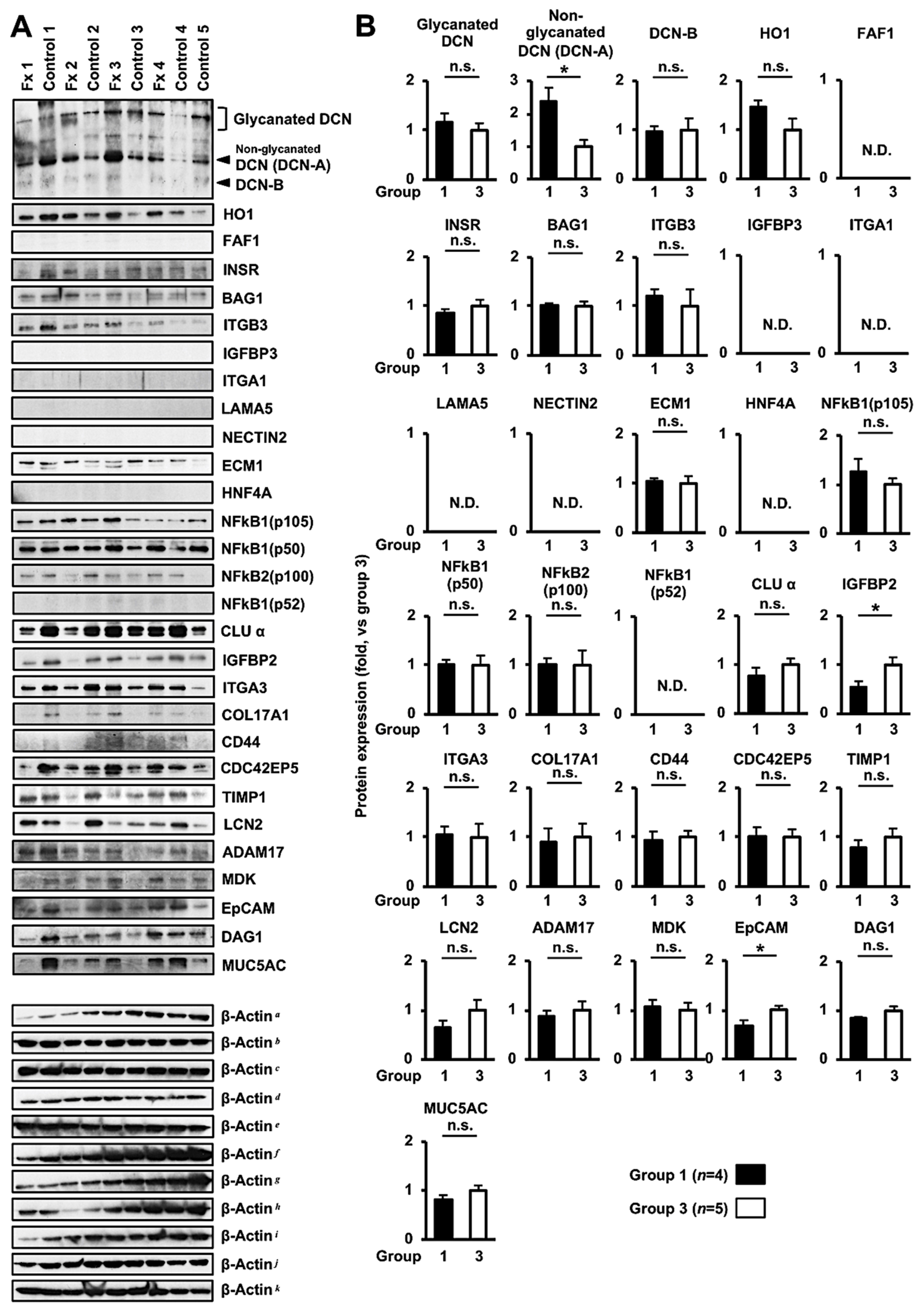
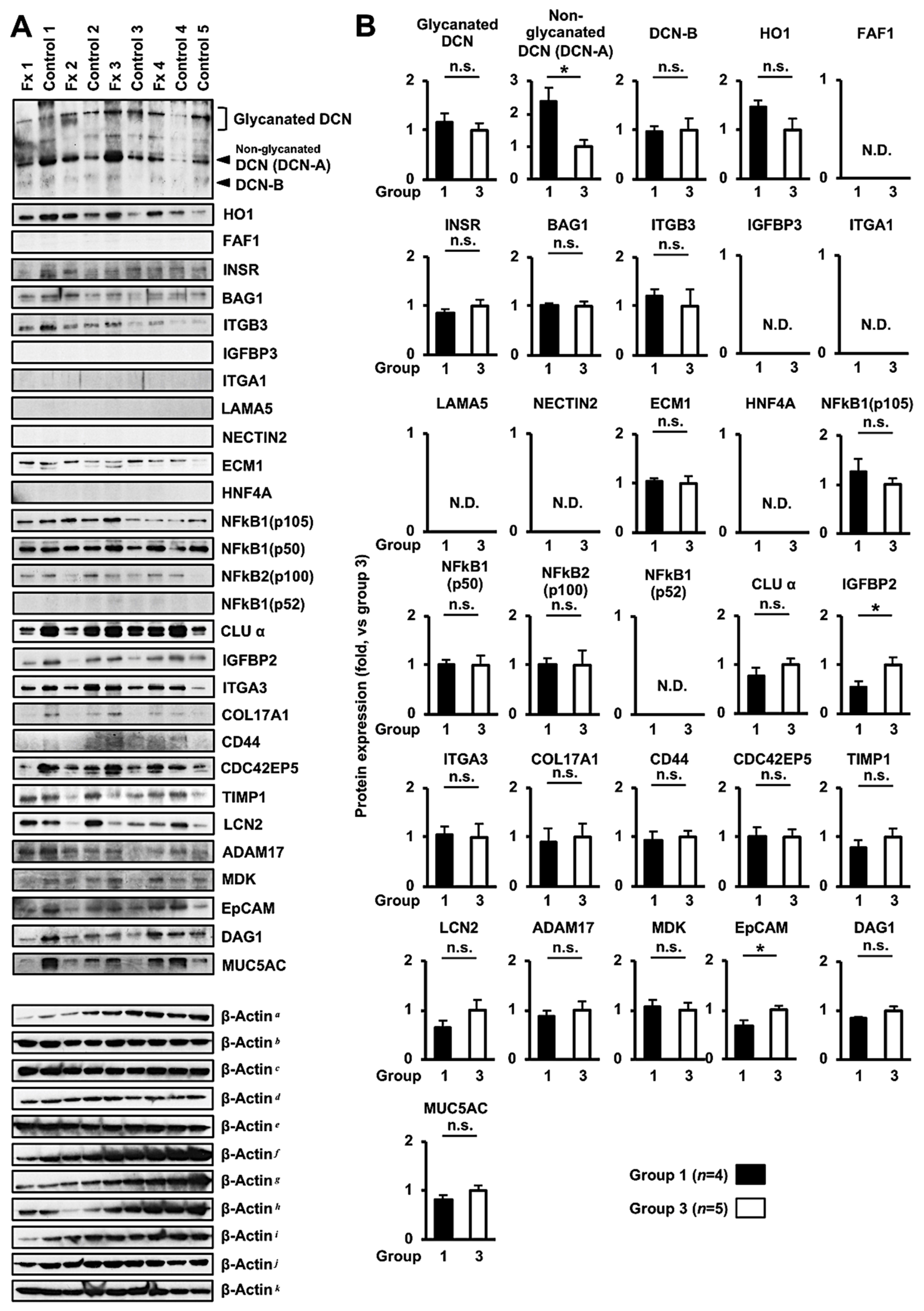
3.3. Comprehensive Proteome Alteration in the Tumor Tissue of PDX Mice after Fx Administration
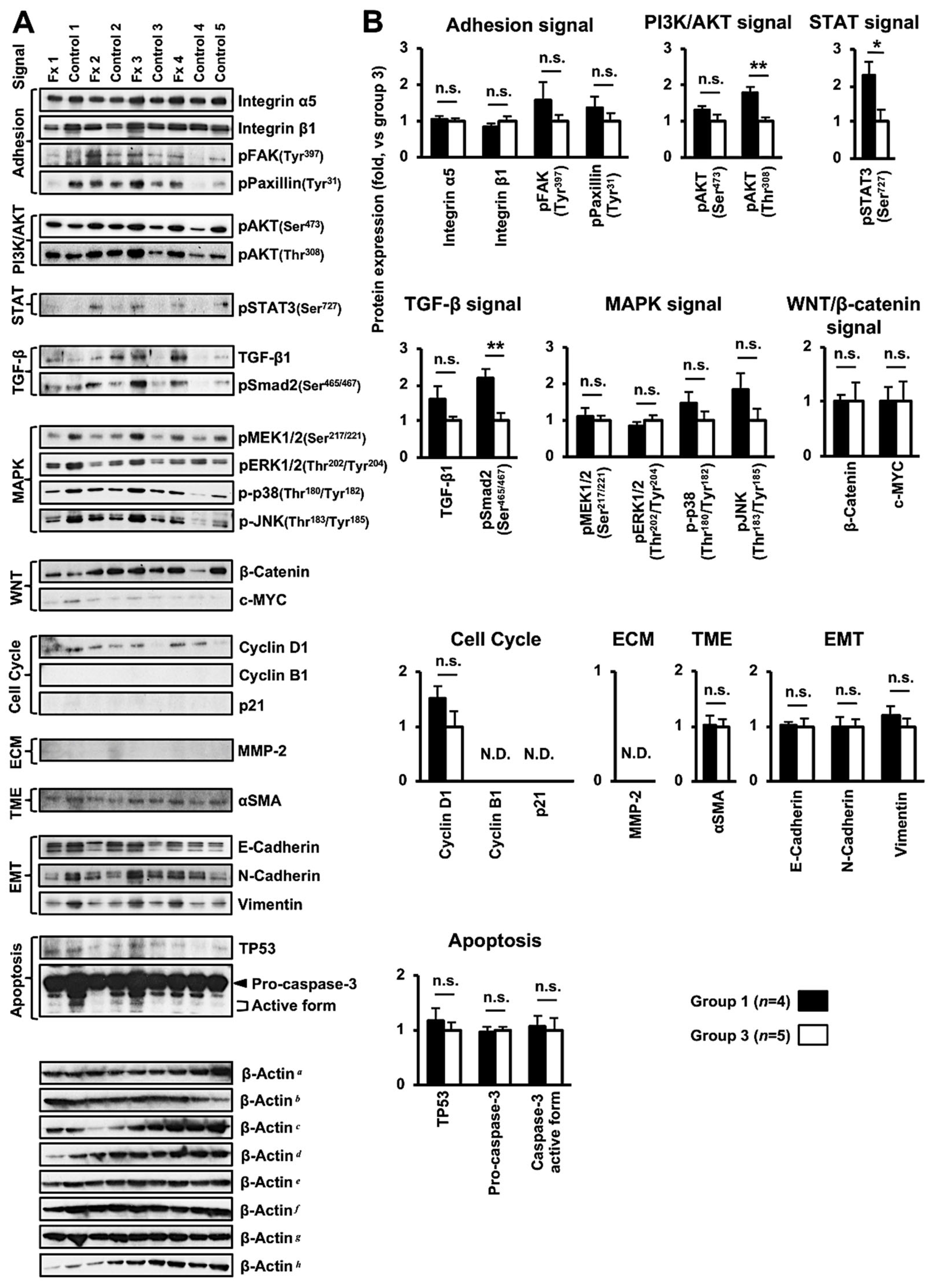
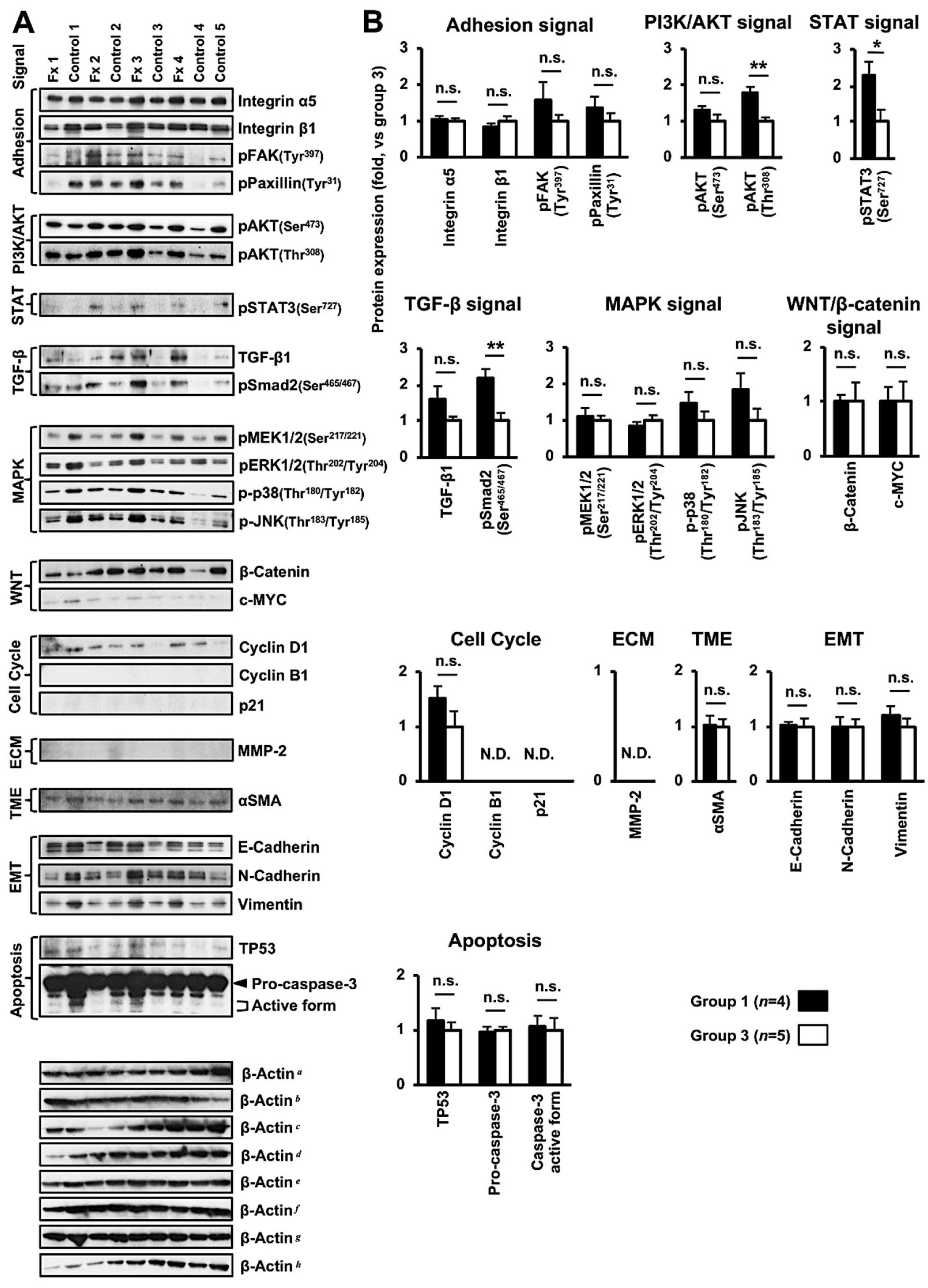
3.4. Altered Protein Expression or Activation in the Tumor Tissues of PDX Mice after Fx Administration Based on Proteome Analyses
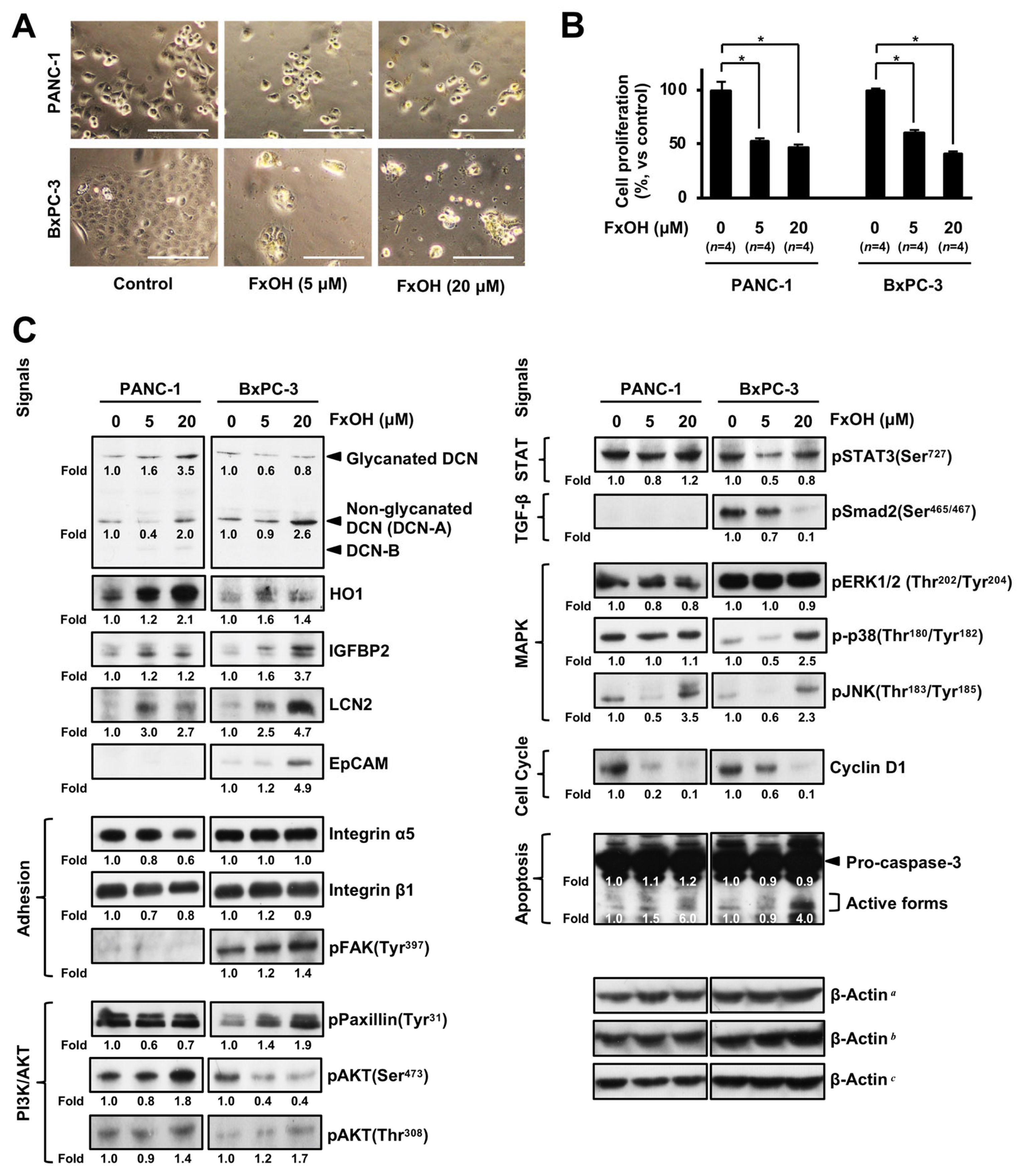
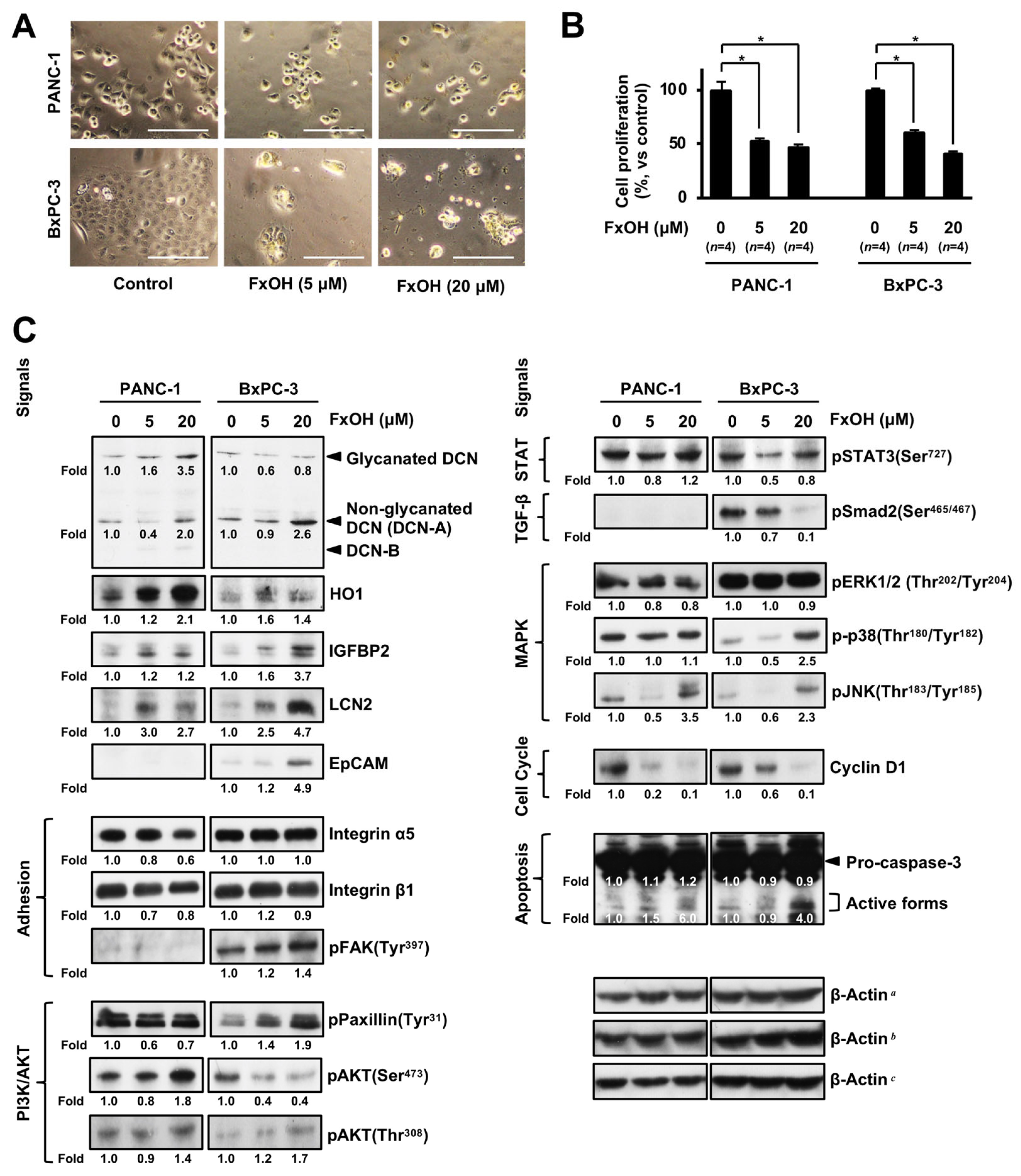
3.5. Altered Protein Expression or Activation in FxOH-Treated Human PC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA N. Open 2021, 4, e214708. [Google Scholar] [CrossRef]
- American Cancer Society. Pancreatic Cancer, American Cancer Society: Atlanta, GA, USA, 2021. Available online: https://www.cancer.org/cancer/pancreatic-cancer.html (accessed on 18 March 2023).
- World Cancer Research Fund; American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Pancretic Cancer 2012. Revised 2018. Available online: https//www.wcrf.org (accessed on 18 March 2023).
- Pelosi, E.; Castelli, G.; Testa, U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 2017, 5, 65. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and Invasive Ductal Pancreatic Cancer and Its Early Detection in The Mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Ijichi, H.; Chytil, A.; Gorska, E.; Aakre, M.E.; Fujitani, Y.; Fujitani, S.; Wright, C.V.E.; Moses, H.L. Aggressive Pancreatic Ductal Adenocarcinoma in Mice Caused by Pancreas-specific Blockade of Transforming Growth Factor-beta Signaling in Cooperation with Active Kras Expression. Genes Dev. 2006, 20, 3147–3160. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Takahashi, M.; Hori, M.; Mutoh, M.; Wakabayashi, K.; Nakagama, H. Experimental Animal Models of Pancreatic Carcinogenesis for Prevention Studies and Their Relevance to Human Disease. Cancers 2011, 3, 582–602. [Google Scholar] [CrossRef]
- Hou, X.; Du, C.; Lu, L.; Yuan, S.; Zhan, M.; You, P.; Du, H. Opportunities and Challenges of Patient-derived Models in Cancer Research: Patient-derived Xenografts, Patient-derived Organoid and Patient-derived Cells. World J. Surg. Oncol. 2022, 20, 37. [Google Scholar] [CrossRef]
- Pan, B.; Wei, X.; Xu, X. Patient-derived Xenograft Models in Hepatopancreatobiliary Cancer. Cancer Cell Int. 2022, 22, 41. [Google Scholar] [CrossRef]
- Izumchenko, E.; Meir, J.; Bedi, A.; Wysocki, P.T.; Hoque, M.O.; Sidransky, D. Patient-derived Xenografts as Tools in Pharmaceutical Development. Clin. Pharmacol. Ther. 2016, 99, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, W.; Long, Y.; Liu, H.; Cheng, J.; Guo, L.; Li, R.; Meng, C.; Yu, S.; Zhao, Q.; et al. Characterization of Drug Responses of Mini Patient-derived Xenografts in Mice for Predicting Cancer Patient Clinical Therapeutic Response. Cancer Commun. 2018, 38, 60. [Google Scholar] [CrossRef]
- Tavares, R.S.N.; Maria-Engler, S.S.; Colepicolo, P.; Debonsi, H.M.; Schäfer-Korting, M.; Marx, U.; Gaspar, L.R.; Zoschke, C. Skin Irritation Testing Beyond Tissue Viability: Fucoxanthin Effects on Inflammation, Homeostasis, and Metabolism. Pharmaceutics 2020, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Beppu, F.; Niwano, Y.; Tsukui, T.; Hosokawa, M.; Miyashita, K. Single and Repeated Oral Dose Toxicity Study of Fucoxanthin (FX), A Marine Carotenoid, in Mice. J. Toxicol. Sci. 2009, 34, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Iio, K.; Okada, Y.; Ishikura, M. Single and 13-week Oral Toxicity Study of Fucoxanthin Oil from Microalgae in Rats. Sokuhin Eiseigaku Zasshi 2011, 52, 183–189. [Google Scholar] [CrossRef]
- Hitoe, S.; Shimoda, H. Seaweed Fucoxanthin Supplementation Improves Obesity Parameters in Mild Obese Japanese Subjects. Funct. Foods Health Dis. 2017, 7, 246–262. [Google Scholar] [CrossRef]
- Mikami, N.; Hosokawa, M.; Miyashita, K.; Sohma, H.; Ito, Y.M.; Kokai, Y. Reduction of HbA1c Levels by Fucoxanthin-enriched Akamoku Oil Possibly Involves the Thrifty Allele of Uncoupling Protein 1 (UCP1): A Randomised Controlled Trial in Normal-Weight and Obese Japanese Adults. J. Nutr. Sci. 2017, 6, e5. [Google Scholar] [CrossRef]
- Adibov, M.; Ramazanov, Z.; Seifulla, R.; Grachev, S. The Effects of Xanthigen in The Weight Management of Obese Premenopausal Women with Non-alcoholic Fatty Liver Disease and Normal Liver Fat. Diabetes Obes. Metab. 2010, 12, 72–81. [Google Scholar] [CrossRef]
- Hashimoto, T.; Ozaki, Y.; Mizuno, M.; Yoshida, M.; Nishitani, Y.; Azuma, T.; Komoto, A.; Maoka, T.; Tanino, Y.; Kanazawa, K. Pharmacokinetics of Fucoxanthinol in Human Plasma after the Oral Administration of Kombu Extract. Br. J. Nutr. 2012, 107, 1566–1569. [Google Scholar] [CrossRef]
- Yonekura, L.; Kobayashi, M.; Terasaki, M.; Nagao, A. Keto-carotenoids Are the Major Metabolites of Dietary Lutein and Fucoxanthin in Mouse Tissues. J. Nutr. 2010, 140, 1824–1831. [Google Scholar] [CrossRef]
- Terasaki, M.; Kubota, A.; Kojima, H.; Maeda, H.; Miyashita, K.; Kawagoe, C.; Mutoh, M.; Tanaka, T. Fucoxanthin and Colorectal Cancer Prevention. Cancers 2021, 13, 2379. [Google Scholar] [CrossRef] [PubMed]
- Nishino, H.; Murakoshi, M.; Tokuda, H.; Satomi, Y. Cancer Prevention by Carotenoids. Arch. Biochem. Biophys. 2009, 483, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Mei, C.; Zhou, S.; Zhu, L.; Ming, J.; Zeng, F.; Xu, R. Antitumor Effects of Laminaria Extract Fucoxanthin on Lung Cancer. Mar. Drugs 2017, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, A.; Wagatsuma, M.; Murase, W.; Kubota, A.; Kojima, H.; Ohta, T.; Hamada, J.; Maeda, H.; Terasaki, M. Fucoxanthinol Promotes Apoptosis in MCF-7 and MDA-MB-231 Cells by Attenuating Laminins-Integrins Axis. Onco 2022, 2, 145–163. [Google Scholar] [CrossRef]
- Terasaki, M.; Inoue, T.; Murase, W.; Kubota, A.; Kojima, H.; Kojoma, M.; Ohta, T.; Maeda, H.; Miyashita, K.; Mutoh, M.; et al. A Fucoxanthinol Induces Apoptosis in a Pancreatic Intraepithelial Neoplasia Cell. Cancer Genom. Proteom. 2021, 18, 133–146. [Google Scholar] [CrossRef]
- Terasaki, M.; Nishizaka, Y.; Murase, W.; Kubota, A.; Kojima, H.; Kojoma, M.; Tanaka, T.; Maeda, H.; Miyashita, K.; Mutoh, M.; et al. Effect of Fucoxanthinol on Pancreatic Ductal Adenocarcinoma Cells From An N-Nitrosobis(2-oxopropyl)amine-initiated Syrian Golden Hamster Pancreatic Carcinogenesis Model. Cancer Genom. Proteom. 2021, 18 (Suppl. 3), 407–423. [Google Scholar] [CrossRef]
- Murase, W.; Kamakura, Y.; Kawakami, S.; Yasuda, A.; Wagatsuma, M.; Kubota, A.; Kojima, H.; Ohta, T.; Takahashi, M.; Mutoh, M.; et al. Fucoxanthin Prevents Pancreatic Tumorigenesis in C57BL/6J Mice That Received Allogenic and Orthotopic Transplants of Cancer Cells. Int. J. Mol. Sci. 2021, 22, 13620. [Google Scholar] [CrossRef]
- Lu, J.; Wu, X.J.; Hassouna, A.; Wang, K.S.; Li, Y.; Feng, T.; Zhao, Y.; Jin, M.; Zhang, B.; Ying, T.; et al. Gemcitabine-fucoxanthin Combination in Human Pancreatic Cancer Cells. Biomed. Rep. 2023, 19, 46. [Google Scholar] [CrossRef]
- Yagishita, S.; Kato, K.; Takahashi, M.; Imai, T.; Yatabe, Y.; Kuwata, T.; Suzuki, M.; Ochiai, A.; Ohtsu, A.; Shimada, K.; et al. Characterization of The Large-scale Japanese Patient-derived Xenograft (J-PDX) library. Cancer Sci. 2021, 112, 2454–2466. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome Sequencing of Pancreatic Cancer Defines Genetic Diversity and Therapeutic Targets. Nature Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A Simple Practice Guide for Dose Conversion between Animals and Human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; U.S. Food & Drug Administration: Rockville, MD, USA, 2005. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers (accessed on 27 March 2023).
- Diehl, V.; Huber, L.S.; Trebicka, J.; Wygrecka, M.; Lozzo, R.V.; Schaefer, L. The Role of Decorin and Biglycan Signaling in Tumorigenesis. Front. Oncol. 2021, 11, 801801. [Google Scholar] [CrossRef] [PubMed]
- Sainio, A.O.; Järveläinen, H.T. Decorin-mediated Oncosuppression—A Potential Future Adjuvant Therapy for Human Epithelial Cancers. Br. J. Pharmacol. 2019, 176, 5–15. [Google Scholar] [CrossRef]
- Buraschi, S.; Neill, T.; Owens, R.T.; Iniguez, L.A.; Purkins, G.; Vadigepalli, R.; Evans, B.; Schaefer, L.; Peiper, S.C.; Wang, Z.X.; et al. Decorin Protein Core Affects the Global Gene Expression Profile of the Tumor Microenvironment in a Triple-Negative Orthotopic Breast Carcinoma Xenograft Model. PLoS ONE 2012, 7, e45559. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, C.; Gao, H.; Zhou, C.; Qin, W.; Wang, J.; Meng, L.; Wang, H.; Ren, Q.; Zhang, Y. Study on the expression profile and role of decorin in the progression of pancreatic cancer. Aging 2021, 13, 14989–14998. [Google Scholar] [CrossRef]
- Ahmad, I.M.; Dafferner, A.J.; O’Connell, K.A.; Mehla, K.; Britigan, B.E.; Hollingsworth, M.A.; Abdalla, M.Y. Heme Oxygenase-1 Inhibition Potentiates the Effects of Nab-Paclitaxel-Gemcitabine and Modulates the Tumor Microenvironment in Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 2264. [Google Scholar] [CrossRef]
- Kim, E.J.; Kim, Y.J.; Lee, H.I.; Jeong, S.H.; Nam, H.J.; Cho, J.H. NRF2 Knockdown Resensitizes 5-Fluorouracil-Resistant Pancreatic Cancer Cells by Suppressing HO-1 and ABCG2 Expression. Int. J. Mol. Sci. 2020, 21, 4646. [Google Scholar] [CrossRef]
- Nuhn, P.; Künzli, B.M.; Hennig, R.; Mitkus, T.; Ramanauskas, T.; Nobiling, R.; Meuer, S.C.; Friess, H.; Berberat, P.O. Heme Oxygenase-1 and Its Metabolites Affect Pancreatic Tumor Growth In Vivo. Mol. Cancer 2009, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Forbes, M.E.; Fuller, G.N.; Li, J.; Yang, X.; Zhang, W. IGFBP2: Integrative Hub of Developmental and Oncogenic Signaling Network. Oncogene 2020, 39, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Radhakrishnan, P. Role of Tumor and Stroma-Derived IGF/IGFBPs in Pancreatic Cancer. Cancers 2020, 12, 1228. [Google Scholar] [CrossRef] [PubMed]
- Gumpper, K.; Dangel, A.W.; Pita-Grisanti, V.; Krishna, S.G.; Lara, L.F.; Mase, T.; Papachristou, G.I.; Conwell, D.L.; Hart, P.A.; Cruz-Monserrate, Z. Lipocalin-2 Expression and Function in Pancreatic Diseases. Pancreatology 2020, 20, 419–424. [Google Scholar] [CrossRef]
- Gomez-Chou, S.B.; Swidnicka-Siergiejko, A.K.; Badi, N.; Chavez-Tomar, M.; Lesinski, G.B.; Bekaii-Saab, T.; Farren, M.R.; Mace, T.A.; Schmidt, C.; Liu, Y.; et al. Lipocalin-2 Promotes Pancreatic Ductal Adenocarcinoma by Regulating Inflammation in the Tumor Microenvironment. Cancer Res. 2017, 77, 2647–2660. [Google Scholar] [CrossRef]
- Barzaman, K.; Vafaei, R.; Samadi, M.; Kazemi, M.H.; Hosseinzadeh, A.; Merikhian, P.; Moradi-Kalbolandi, S.; Eisavand, M.R.; Dinvari, H.; Farahmand, L. Anti-cancer Therapeutic Strategies Based on HGF/MET, EpCAM, and Tumor-stromal Cross Talk. Cancer Int. 2022, 22, 259. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Budzinska, A.; Mojzych, M.; Kontek, R. Metastasis and MAPK Pathways. Int. J. Mol. Sci. 2022, 23, 3847. [Google Scholar] [CrossRef]
- Li, Y.; Hong, J.W.; Oh, J.E.; Yoon, A.R.; Yun, C.O. Potent Antitumor Effect of Tumor Microenvironment-targeted Oncolytic Adenovirus Against Desmoplastic Pancreatic Cancer. Int. J. Cancer 2018, 142, 392–413. [Google Scholar] [CrossRef]
- Liu, H.; Li, L.; Chen, H.; Kong, R.; Pan, S.; Hu, J.; Wang, Y.; Li, Y.; Sun, B. Silencing IGFBP-2 Decreases Pancreatic Cancer Metastasis and Enhances Chemotherapeutic Sensitivity. Oncotarget 2017, 8, 61674–616866. [Google Scholar] [CrossRef]
- Leung, L.; Radulovich, N.; Zhu, C.Q.; Organ, S.; Bandarchi, B.; Pintille, M.; To, C.; Panchal, D.; Tsao, M.S. Lipocalin2 Promotes Invasion, Tumorigenicity and Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. PLoS ONE 2012, 7, e46677. [Google Scholar] [CrossRef]
- Salnikov, A.V.; Groth, A.; Apel, A.; Kallifatidis, G.; Beckermann, B.M.; Khamidjanov, A.; Ryschich, E.; Büchler, M.W.; Herr, I.; Moldenhauer, G. Targeting of Cancer Stem Cell Marker EpCAM by Bispecific Antibody EpCAMxCD3 Inhibits Pancreatic Carcinoma. J. Cell Mol. Med. 2009, 13, 4023–4033. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Song, M.H.; Oh, J.W.; Keum, Y.S.; Saini, R.K. Pro-Oxidant Actions of Carotenoids in Triggering Apoptosis of Cancer Cells: A Review of Emerging Evidence. Antioxidants 2020, 9, 532. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, V.; Berginc, G.; Volavsek, M.; Stor, Z.; Rems, M.; Glavac, D. Presence of Activating KRAS Mutations Correlates Significantly with Expression of Tumour Suppressor Genes DCN and TPMI in Colorectal Cancer. BMC Cancer 2009, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Dunlap, S.M.; Celestino, J.; Wang, H.; Jiang, R.; Holland, E.C.; Fuller, G.N.; Zhang, W. Insulin-like growth factor binding protein 2 promotes glioma development and progression. Proc. Natl. Acad. Sci. USA 2007, 104, 11736–11741. [Google Scholar] [CrossRef]
- Livshits, G.; Alonso-Curbelo, D.; Morris, J.P., 4th; Koche, R.; Saborowski, M.; Wilkinson, J.E.; Lowe, S.W. Arid1a Restrains Kras-dependent Changes in Acinar Cell Identity. Elife 2018, 7, e35216. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Characteristics |
|---|---|
| Registration number | X130068 |
| Race | Japanese |
| Age (years) | Early 70s |
| Gender | Male |
| Location of sample collection | Pancreas |
| Disease | Primary pancreatic cancer without recurrence |
| Pathological type | Adenocarcinoma |
| TNM status | T4N1M1 |
| Clinical stage | Stage IV |
| Chemotherapy, radiotherapy and immunotherapy | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terasaki, M.; Suzuki, S.; Tanaka, T.; Maeda, H.; Shibata, M.; Miyashita, K.; Kuramitsu, Y.; Hamada, J.; Ohta, T.; Yagishita, S.; et al. Anticancer Effects of Fucoxanthin in a PDX Model of Advanced Stage Pancreatic Cancer with Alteration of Several Multifunctional Molecules. Onco 2023, 3, 217-236. https://doi.org/10.3390/onco3040016
Terasaki M, Suzuki S, Tanaka T, Maeda H, Shibata M, Miyashita K, Kuramitsu Y, Hamada J, Ohta T, Yagishita S, et al. Anticancer Effects of Fucoxanthin in a PDX Model of Advanced Stage Pancreatic Cancer with Alteration of Several Multifunctional Molecules. Onco. 2023; 3(4):217-236. https://doi.org/10.3390/onco3040016
Chicago/Turabian StyleTerasaki, Masaru, Sally Suzuki, Takuji Tanaka, Hayato Maeda, Masaki Shibata, Kazuo Miyashita, Yasuhiro Kuramitsu, Junichi Hamada, Tohru Ohta, Shigehiro Yagishita, and et al. 2023. "Anticancer Effects of Fucoxanthin in a PDX Model of Advanced Stage Pancreatic Cancer with Alteration of Several Multifunctional Molecules" Onco 3, no. 4: 217-236. https://doi.org/10.3390/onco3040016