

Monoclonal Antibody Development for Cancer Treatment Using the Phage Display Library Platform
Abstract
:1. Introduction
2. Overview of Procedures of Phage Display Technology in Monoclonal Antibody Production
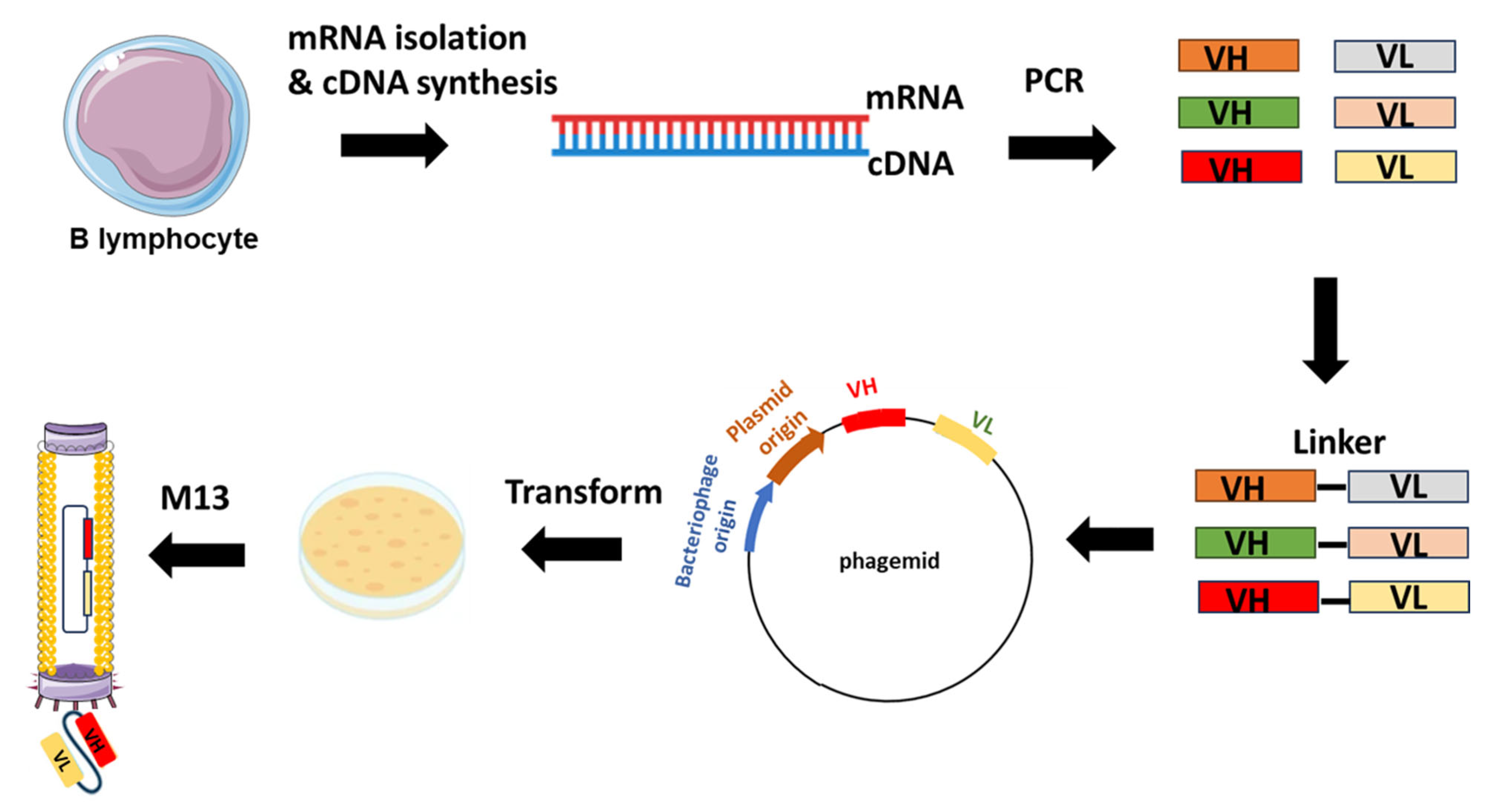
2.1. Library Construction
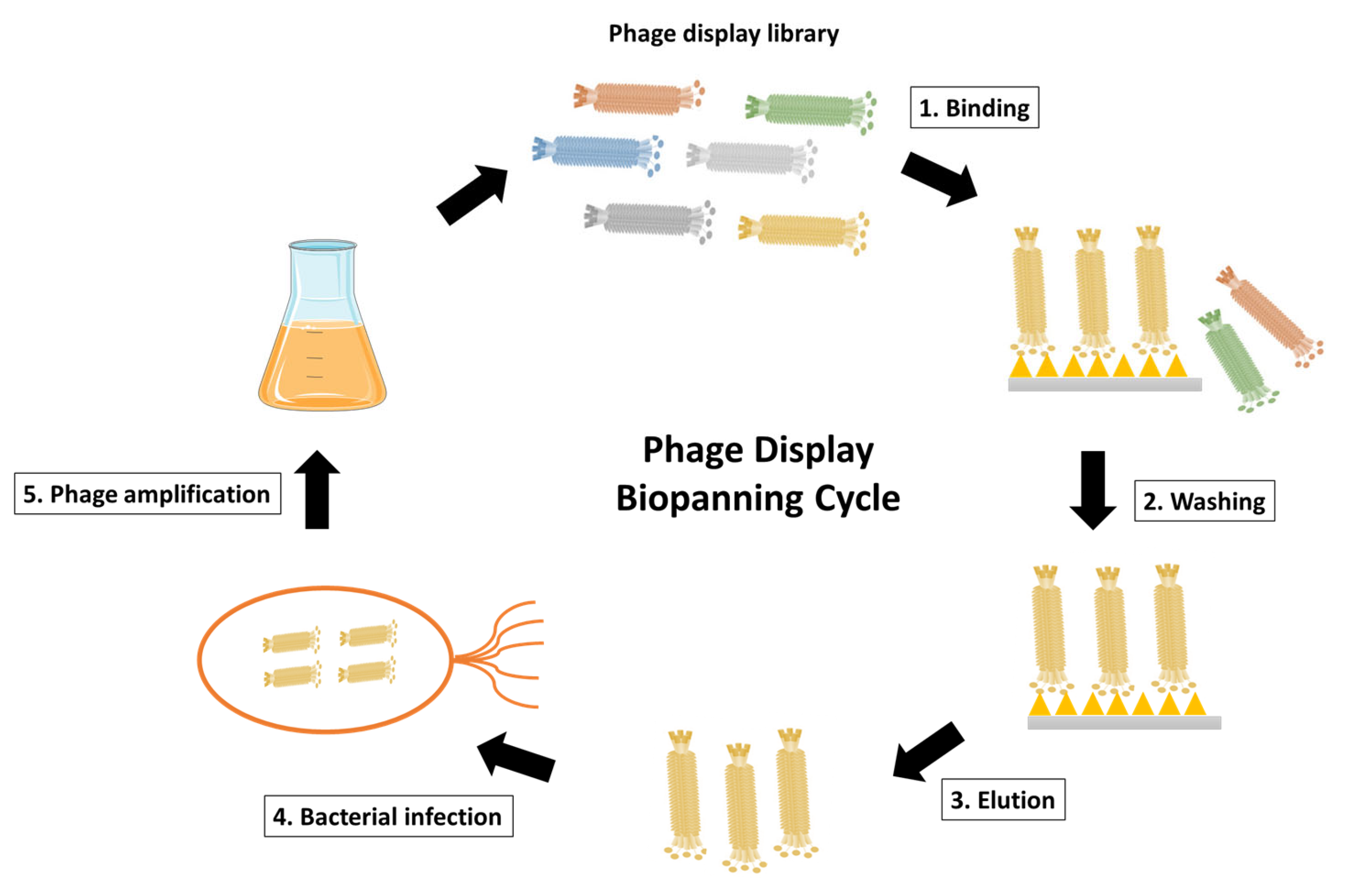
2.2. Biopanning, Elution and Amplification
2.3. DNA Sequencing and Antibody Production
2.4. Characterization and Optimization
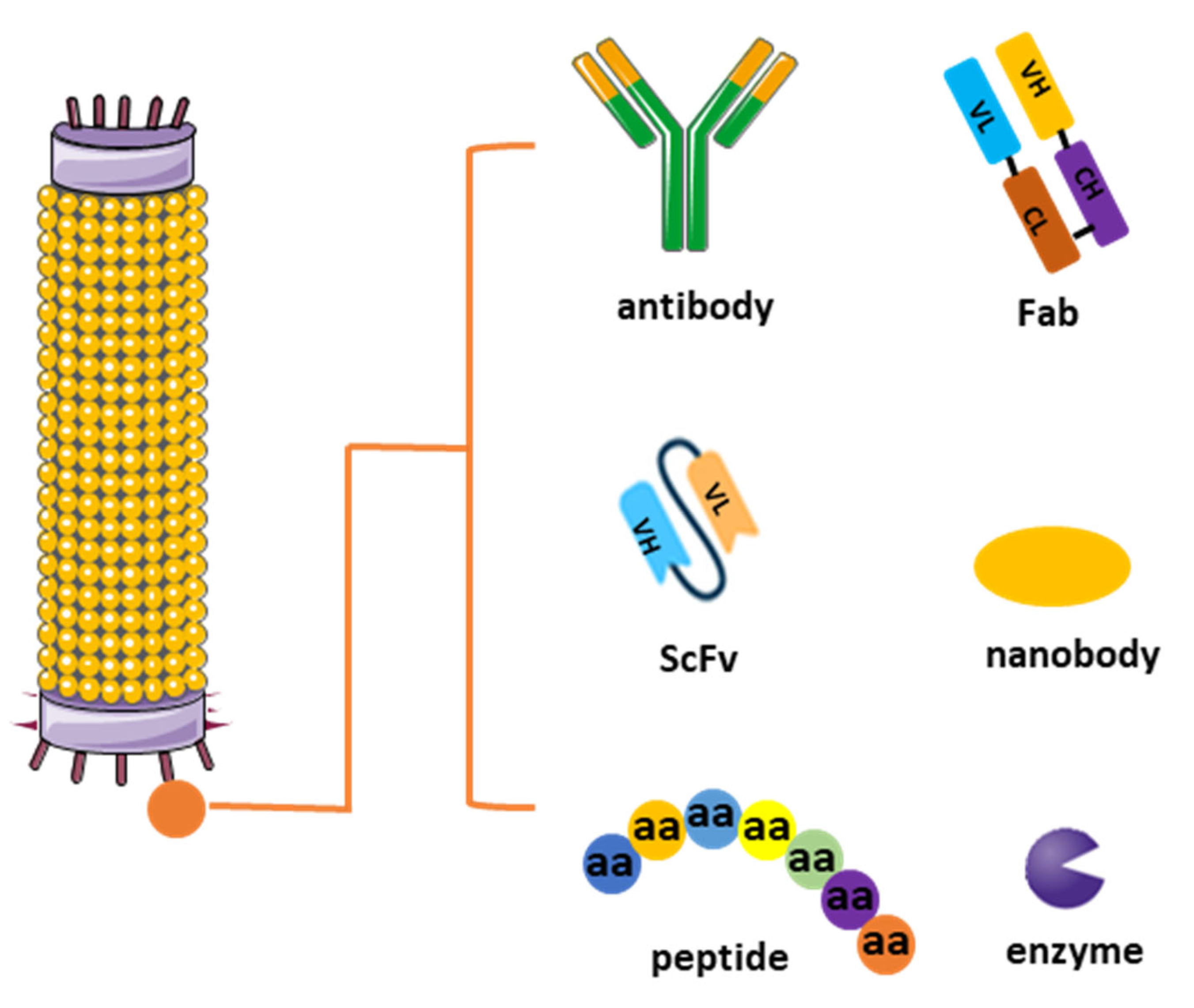
3. Categories of Phage Display Libraries for Antibody Development
4. Phage Display Libraries for Antibody Development for Cancer Treatment
4.1. The Advantages of Using Phage Display Libraries for Antibody Development
4.2. Phage Display-Derived mAbs for Cancer Treatment
4.2.1. Atezolizumab
4.2.2. Avelumab
4.2.3. Moxetumomab Pasudotox-tdfk
4.2.4. Necitumumab
4.2.5. Ramucirumab
5. Discussion of Challenges and Future Prospectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Stanfield, R.L.; Wilson, I.A. Antibody Structure. Microbiol. Spectr. 2014, 2, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.L.; Gilliland, G.L. Engineering antibody therapeutics. Curr. Opin. Struct. Biol. 2016, 38, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Shim, H. Antibody Phage Display. Adv. Exp. Med. Biol. 2017, 1053, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Franca, R.K.A.; Studart, I.C.; Bezerra, M.R.L.; Pontes, L.Q.; Barbosa, A.M.A.; Brigido, M.M.; Furtado, G.P.; Maranhao, A.Q. Progress on Phage Display Technology: Tailoring Antibodies for Cancer Immunotherapy. Viruses 2023, 15, 1903. [Google Scholar] [CrossRef] [PubMed]
- Domm, W.; Brewer, M.; Baker, S.F.; Feng, C.; Martinez-Sobrido, L.; Treanor, J.; Dewhurst, S. Use of bacteriophage particles displaying influenza virus hemagglutinin for the detection of hemagglutination-inhibition antibodies. J. Virol. Methods 2014, 197, 47–50. [Google Scholar] [CrossRef]
- Chikaev, A.N.; Chikaev, A.N.; Rudometov, A.P.; Merkulyeva, Y.A.; Karpenko, L.I. Phage display as a tool for identifying HIV-1 broadly neutralizing antibodies. Vavilovskii Zhurnal Genet. Sel. 2021, 25, 562–572. [Google Scholar] [CrossRef]
- Peltomaa, R.; Benito-Pena, E.; Barderas, R.; Moreno-Bondi, M.C. Phage Display in the Quest for New Selective Recognition Elements for Biosensors. ACS Omega 2019, 4, 11569–11580. [Google Scholar] [CrossRef]
- Jones, M.L.; Alfaleh, M.A.; Kumble, S.; Zhang, S.; Osborne, G.W.; Yeh, M.; Arora, N.; Hou, J.J.; Howard, C.B.; Chin, D.Y.; et al. Targeting membrane proteins for antibody discovery using phage display. Sci. Rep. 2016, 6, 26240. [Google Scholar] [CrossRef]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Paeshuyse, J. Construction of Antibody Phage Libraries and Their Application in Veterinary Immunovirology. Antibodies 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.; Calkosinski, I.; Gamian, A. Phage display--a powerful technique for immunotherapy: 1. Introduction and potential of therapeutic applications. Hum. Vaccin. Immunother. 2012, 8, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Anand, T.; Virmani, N.; Bera, B.C.; Vaid, R.K.; Vashisth, M.; Bardajatya, P.; Kumar, A.; Tripathi, B.N. Phage Display Technique as a Tool for Diagnosis and Antibody Selection for Coronaviruses. Curr. Microbiol. 2021, 78, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Song, E.W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar] [CrossRef]
- Nascimento, A.; Mullerpatan, A.; Azevedo, A.M.; Karande, P.; Cramer, S. Development of phage biopanning strategies to identify affinity peptide ligands for kappa light chain Fab fragments. Biotechnol. Prog. 2019, 35, e2884. [Google Scholar] [CrossRef]
- Tulika, T.; Pedersen, R.W.; Rimbault, C.; Ahmadi, S.; Rivera-de-Torre, E.; Fernandez-Quintero, M.L.; Loeffler, J.R.; Bohn, M.F.; Ljungars, A.; Ledsgaard, L.; et al. Phage display assisted discovery of a pH-dependent anti-alpha-cobratoxin antibody from a natural variable domain library. Protein Sci. 2023, 32, e4821. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Feng, N.; Li, Y.; Gu, J.; Wang, Z. Developability assessment at early-stage discovery to enable development of antibody-derived therapeutics. Antib. Ther. 2023, 6, 13–29. [Google Scholar] [CrossRef]
- Li, W.; Caberoy, N.B. New perspective for phage display as an efficient and versatile technology of functional proteomics. Appl. Microbiol. Biotechnol. 2010, 85, 909–919. [Google Scholar] [CrossRef]
- Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Iorno, N.; Sierro, F.; Christ, D. Selection of human antibody fragments by phage display. Nat. Protoc. 2007, 2, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Prassler, J.; Thiel, S.; Pracht, C.; Polzer, A.; Peters, S.; Bauer, M.; Norenberg, S.; Stark, Y.; Kolln, J.; Popp, A.; et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J. Mol. Biol. 2011, 413, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Gram, H.; Marconi, L.A.; Barbas, C.F., 3rd; Collet, T.A.; Lerner, R.A.; Kang, A.S. In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc. Natl. Acad. Sci. USA 1992, 89, 3576–3580. [Google Scholar] [CrossRef] [PubMed]
- Bao, G.; Tang, M.; Zhao, J.; Zhu, X. Nanobody: A promising toolkit for molecular imaging and disease therapy. EJNMMI Res. 2021, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Ding, Z.; Yang, X.; Zhao, X.; Zhao, M.; Gao, L.; Chen, Q.; Xie, S.; Liu, A.; Yin, S.; et al. Nanobody: A Small Antibody with Big Implications for Tumor Therapeutic Strategy. Int. J. Nanomed. 2021, 16, 2337–2356. [Google Scholar] [CrossRef]
- Weber, M.; Bujak, E.; Putelli, A.; Villa, A.; Matasci, M.; Gualandi, L.; Hemmerle, T.; Wulhfard, S.; Neri, D. A highly functional synthetic phage display library containing over 40 billion human antibody clones. PLoS ONE 2014, 9, e100000. [Google Scholar] [CrossRef]
- Kumar, R.; Parray, H.A.; Shrivastava, T.; Sinha, S.; Luthra, K. Phage display antibody libraries: A robust approach for generation of recombinant human monoclonal antibodies. Int. J. Biol. Macromol. 2019, 135, 907–918. [Google Scholar] [CrossRef]
- Andre, A.S.; Moutinho, I.; Dias, J.N.R.; Aires-da-Silva, F. In vivo Phage Display: A promising selection strategy for the improvement of antibody targeting and drug delivery properties. Front. Microbiol. 2022, 13, 962124. [Google Scholar] [CrossRef]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. MAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.K.; Rahumatullah, A.; Lai, J.Y.; Lim, T.S. Naive Human Antibody Libraries for Infectious Diseases. Adv. Exp. Med. Biol. 2017, 1053, 35–59. [Google Scholar] [CrossRef] [PubMed]
- Braunagel, M.; Little, M. Construction of a semisynthetic antibody library using trinucleotide oligos. Nucleic Acids Res. 1997, 25, 4690–4691. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021, 42, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Ormundo, L.F.; Barreto, C.T.; Tsuruta, L.R. Development of Therapeutic Monoclonal Antibodies for Emerging Arbovirus Infections. Viruses 2023, 15, 2177. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.Z.; Hamaguchi, B.; Braggion, C.; Speciale, E.R.; Cesar, F.B.V.; Soares, G.d.F.d.S.; Osaki, J.H.; Pereira, T.M.; Aguiar, R.B. Hybridoma technology: Is it still useful? Curr. Res. Immunol. 2021, 2, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Callanan, D.; Kunimoto, D.; Maturi, R.K.; Patel, S.S.; Staurenghi, G.; Wolf, S.; Cheetham, J.K.; Hohman, T.C.; Kim, K.; Lopez, F.J.; et al. Double-Masked, Randomized, Phase 2 Evaluation of Abicipar Pegol (an Anti-VEGF DARPin Therapeutic) in Neovascular Age-Related Macular Degeneration. J. Ocul. Pharmacol. Ther. 2018, 34, 700–709. [Google Scholar] [CrossRef]
- Markham, A. Atezolizumab: First Global Approval. Drugs 2016, 76, 1227–1232. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef]
- Ho, M.; Nagata, S.; Pastan, I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 9637–9642. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kussie, P.; Ferguson, K.M. Structural basis for EGF receptor inhibition by the therapeutic antibody IMC-11F8. Structure 2008, 16, 216–227. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Necitumumab: First Global Approval. Drugs 2016, 76, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Shen, J.; Vil, M.D.; Zhang, H.; Jimenez, X.; Bohlen, P.; Witte, L.; Zhu, Z. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J. Biol. Chem. 2003, 278, 43496–43507. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Jimenez, X.; Zhang, H.; Bohlen, P.; Witte, L.; Zhu, Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int. J. Cancer 2002, 97, 393–399. [Google Scholar] [CrossRef]
- Kim, E.S. Avelumab: First Global Approval. Drugs 2017, 77, 929–937. [Google Scholar] [CrossRef]
- Nixon, A.E.; Sexton, D.J.; Ladner, R.C. Drugs derived from phage display: From candidate identification to clinical practice. MAbs 2014, 6, 73–85. [Google Scholar] [CrossRef]
- Kindler, H.L.; Richards, D.A.; Garbo, L.E.; Garon, E.B.; Stephenson, J.J., Jr.; Rocha-Lima, C.M.; Safran, H.; Chan, D.; Kocs, D.M.; Galimi, F.; et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann. Oncol. 2012, 23, 2834–2842. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Capanu, M.; O’Reilly, E.M.; Ma, J.; Chou, J.F.; Gansukh, B.; Shia, J.; Kalin, M.; Katz, S.; Abad, L.; et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J. Hepatol. 2014, 60, 319–324. [Google Scholar] [CrossRef]
- Liu, J.F.; Ray-Coquard, I.; Selle, F.; Poveda, A.M.; Cibula, D.; Hirte, H.; Hilpert, F.; Raspagliesi, F.; Gladieff, L.; Harter, P.; et al. Randomized Phase II Trial of Seribantumab in Combination with Paclitaxel in Patients with Advanced Platinum-Resistant or -Refractory Ovarian Cancer. J. Clin. Oncol. 2016, 34, 4345–4353. [Google Scholar] [CrossRef]
- Younes, A.; Vose, J.M.; Zelenetz, A.D.; Smith, M.R.; Burris, H.A.; Ansell, S.M.; Klein, J.; Halpern, W.; Miceli, R.; Kumm, E.; et al. A Phase 1b/2 trial of mapatumumab in patients with relapsed/refractory non-Hodgkin’s lymphoma. Br. J. Cancer 2010, 103, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Scheulen, M.E.; Dittrich, C.; Obrist, P.; Marschner, N.; Dirix, L.; Schmidt, M.; Ruttinger, D.; Schuler, M.; Reinhardt, C.; et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann. Oncol. 2010, 21, 275–282. [Google Scholar] [CrossRef]
- Dowlati, A.; Vlahovic, G.; Natale, R.B.; Rasmussen, E.; Singh, I.; Hwang, Y.C.; Rossi, J.; Bass, M.B.; Friberg, G.; Pickett, C.A. A Phase I, First-in-Human Study of AMG 780, an Angiopoietin-1 and -2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 4574–4584. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Knost, J.A.; Chiorean, E.G.; Kambhampati, S.R.; Yu, D.; Pytowski, B.; Qin, A.; Kauh, J.S.; O’Neil, B.H. Phase 1 study of the anti-vascular endothelial growth factor receptor 3 monoclonal antibody LY3022856/IMC-3C5 in patients with advanced and refractory solid tumors and advanced colorectal cancer. Cancer Chemother. Pharmacol. 2016, 78, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Patel, M.R.; Sachdev, J.C.; Hart, L.L.; Halama, N.; Ramanathan, R.K.; Sarantopoulos, J.; Volkel, D.; Youssef, A.; de Jong, F.A.; et al. Phase I study of imalumab (BAX69), a fully human recombinant antioxidized macrophage migration inhibitory factor antibody in advanced solid tumours. Br. J. Clin. Pharmacol. 2020, 86, 1836–1848. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Engelhardt, M.; Blank, A.; Goldschmidt, H.; Agis, H.; Blau, I.W.; Einsele, H.; Ferstl, B.; Schub, N.; Rollig, C.; et al. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: A first-in-human, multicentre, phase 1-2a trial. Lancet Haematol. 2020, 7, e381–e394. [Google Scholar] [CrossRef]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab ravtansine: A novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef]
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Investig. New Drugs 2019, 37, 722–730. [Google Scholar] [CrossRef]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L.; et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Mateos, C.; Alcaraz-Serna, A.; Somovilla-Crespo, B.; Munoz-Calleja, C. Monoclonal Antibody Therapies for Hematological Malignancies: Not Just Lineage-Specific Targets. Front. Immunol. 2017, 8, 1936. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jin, J.; Guo, D.; Tao, Z.; Hu, X. Immune Checkpoint Inhibitors Combined with Targeted Therapy: The Recent Advances and Future Potentials. Cancers 2023, 15, 2858. [Google Scholar] [CrossRef]
- Pelizzaro, F.; Farinati, F.; Trevisani, F. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance. Biomedicines 2023, 11, 1020. [Google Scholar] [CrossRef] [PubMed]
- Uchimiak, K.; Badowska-Kozakiewicz, A.M.; Sobiborowicz-Sadowska, A.; Deptala, A. Current State of Knowledge on the Immune Checkpoint Inhibitors in Triple-Negative Breast Cancer Treatment: Approaches, Efficacy, and Challenges. Clin. Med. Insights Oncol. 2022, 16, 11795549221099869. [Google Scholar] [CrossRef]
- Zakharia, Y.; Huynh, L.; Du, S.; Chang, R.; Pi, S.; Sundaresan, S.; Duh, M.S.; Zanotti, G.; Thomaidou, D. Impact of Therapy Management on Axitinib-Related Adverse Events in Patients with Advanced Renal Cell Carcinoma Receiving First-Line Axitinib + Checkpoint Inhibitor. Clin. Genitourin. Cancer 2023, 21, e343–e351. [Google Scholar] [CrossRef]
- Conforti, F.; Zucali, P.A.; Pala, L.; Catania, C.; Bagnardi, V.; Sala, I.; Della Vigna, P.; Perrino, M.; Zagami, P.; Corti, C.; et al. Avelumab plus axitinib in unresectable or metastatic type B3 thymomas and thymic carcinomas (CAVEATT): A single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022, 23, 1287–1296. [Google Scholar] [CrossRef]
- Nobre, C.F.; Newman, M.J.; DeLisa, A.; Newman, P. Moxetumomab pasudotox-tdfk for relapsed/refractory hairy cell leukemia: A review of clinical considerations. Cancer Chemother. Pharmacol. 2019, 84, 255–263. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Robak, T.; le Coutre, P.D.; Gjertsen, B.T.; Troussard, X.; Roboz, G.J.; Karlin, L.; et al. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): Long-term follow-up from the pivotal trial. J. Hematol. Oncol. 2021, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Bhojwani, D.; August, K.; Baruchel, A.; Bertrand, Y.; Boklan, J.; Dalla-Pozza, L.; Dennis, R.; Hijiya, N.; Locatelli, F.; et al. Results from an international phase 2 study of the anti-CD22 immunotoxin moxetumomab pasudotox in relapsed or refractory childhood B-lineage acute lymphoblastic leukemia. Pediatr. Blood Cancer 2020, 67, e28112. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Zhang, H.; Koo, H.; Tonra, J.; Balderes, P.; Prewett, M.; Corcoran, E.; Mangalampalli, V.; Bassi, R.; Anselma, D.; et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J. Biol. Chem. 2005, 280, 19665–19672. [Google Scholar] [CrossRef] [PubMed]
- Fala, L. Portrazza (Necitumumab), an IgG1 Monoclonal Antibody, FDA Approved for Advanced Squamous Non-Small-Cell Lung Cancer. Am. Health Drug Benefits 2016, 9, 119–122. [Google Scholar]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Ciuleanu, T.E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.; et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): A multicentre, double-blind, randomised phase 3 trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Singal, A.G.; Villanueva, A.; Finn, R.S.; Kudo, M.; Galle, P.R.; Ikeda, M.; Callies, S.; McGrath, L.M.; Wang, C.; et al. Prognostic and Predictive Factors in Patients with Advanced HCC and Elevated Alpha-Fetoprotein Treated with Ramucirumab in Two Randomized Phase III Trials. Clin. Cancer Res. 2022, 28, 2297–2305. [Google Scholar] [CrossRef]
- Debeuckelaere, C.; Murgioni, S.; Lonardi, S.; Girardi, N.; Alberti, G.; Fano, C.; Gallimberti, S.; Magro, C.; Ahcene-Djaballah, S.; Daniel, F.; et al. Ramucirumab: The long and winding road toward being an option for mCRC treatment. Expert Opin. Biol. Ther. 2019, 19, 399–409. [Google Scholar] [CrossRef]
- Shaib, W.L.; Manali, R.; Liu, Y.; El-Rayes, B.; Loehrer, P.; O’Neil, B.; Cohen, S.; Khair, T.; Robin, E.; Huyck, T.; et al. Phase II randomised, double-blind study of mFOLFIRINOX plus ramucirumab versus mFOLFIRINOX plus placebo in advanced pancreatic cancer patients (HCRN GI14-198). Eur. J. Cancer 2023, 189, 112847. [Google Scholar] [CrossRef]
- Mackey, J.R.; Ramos-Vazquez, M.; Lipatov, O.; McCarthy, N.; Krasnozhon, D.; Semiglazov, V.; Manikhas, A.; Gelmon, K.A.; Konecny, G.E.; Webster, M.; et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J. Clin. Oncol. 2015, 33, 141–148. [Google Scholar] [CrossRef]
- de Wit, R.; Powles, T.; Castellano, D.; Necchi, A.; Lee, J.L.; van der Heijden, M.S.; Matsubara, N.; Bamias, A.; Flechon, A.; Sternberg, C.N.; et al. Exposure-response relationship of ramucirumab in RANGE, a randomized phase III trial in advanced urothelial carcinoma refractory to platinum therapy. Br. J. Clin. Pharmacol. 2022, 88, 3182–3192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kordish, D.H.; Suryawanshi, Y.R.; Eversole, R.R.; Kohler, S.; Mackenzie, C.D.; Essani, K. Oncolytic Tanapoxvirus Expressing Interleukin-2 is Capable of Inducing the Regression of Human Melanoma Tumors in the Absence of T Cells. Curr. Cancer Drug Targets 2018, 18, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, T.; Anderson, A.; Lee, V.; Szymura, S.; Dong, Z.; Kuang, B.; Oh, E.; Liu, J.; Neelapu, S.S.; et al. Immortalized B Cells Transfected with mRNA of Antigen Fused to MITD (IBMAM): An Effective Tool for Antigen-Specific T-Cell Expansion and TCR Validation. Biomedicines 2023, 11, 796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Jou, T.H.; Hsin, J.; Wang, Z.; Huang, K.; Ye, J.; Yin, H.; Xing, Y. Talimogene Laherparepvec (T-VEC): A Review of the Recent Advances in Cancer Therapy. J. Clin. Med. 2023, 12, 1098. [Google Scholar] [CrossRef]
- Greenwood, J.; Willis, A.E.; Perham, R.N. Multiple display of foreign peptides on a filamentous bacteriophage. Peptides from Plasmodium falciparum circumsporozoite protein as antigens. J. Mol. Biol. 1991, 220, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Lamboy, J.A.; Arter, J.A.; Knopp, K.A.; Der, D.; Overstreet, C.M.; Palermo, E.F.; Urakami, H.; Yu, T.B.; Tezgel, O.; Tew, G.N.; et al. Phage wrapping with cationic polymers eliminates nonspecific binding between M13 phage and high pI target proteins. J Am. Chem. Soc. 2009, 131, 16454–16460. [Google Scholar] [CrossRef]
- Akbar, R.; Bashour, H.; Rawat, P.; Robert, P.A.; Smorodina, E.; Cotet, T.S.; Flem-Karlsen, K.; Frank, R.; Mehta, B.B.; Vu, M.H.; et al. Progress and challenges for the machine learning-based design of fit-for-purpose monoclonal antibodies. MAbs 2022, 14, 2008790. [Google Scholar] [CrossRef]
- Zambrano, N.; Froechlich, G.; Lazarevic, D.; Passariello, M.; Nicosia, A.; De Lorenzo, C.; Morelli, M.J.; Sasso, E. High-Throughput Monoclonal Antibody Discovery from Phage Libraries: Challenging the Current Preclinical Pipeline to Keep the Pace with the Increasing mAb Demand. Cancers 2022, 14, 1325. [Google Scholar] [CrossRef]
- Islam, M.S.; Fan, J.; Pan, F. The power of phages: Revolutionizing cancer treatment. Front. Oncol. 2023, 13, 1290296. [Google Scholar] [CrossRef]
- Brown, K.C. Peptidic tumor targeting agents: The road from phage display peptide selections to clinical applications. Curr. Pharm. Des. 2010, 16, 1040–1054. [Google Scholar] [CrossRef]
- Liu, J.K.; Lubelski, D.; Schonberg, D.L.; Wu, Q.; Hale, J.S.; Flavahan, W.A.; Mulkearns-Hubert, E.E.; Man, J.; Hjelmeland, A.B.; Yu, J.; et al. Phage display discovery of novel molecular targets in glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 1325–1339. [Google Scholar] [CrossRef]
- de Wildt, R.M.; Mundy, C.R.; Gorick, B.D.; Tomlinson, I.M. Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat. Biotechnol. 2000, 18, 989–994. [Google Scholar] [CrossRef]
- Munke, A.; Persson, J.; Weiffert, T.; De Genst, E.; Meisl, G.; Arosio, P.; Carnerup, A.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Phage display and kinetic selection of antibodies that specifically inhibit amyloid self-replication. Proc. Natl. Acad. Sci. USA 2017, 114, 6444–6449. [Google Scholar] [CrossRef]
- Rader, C.; Cheresh, D.A.; Barbas, C.F., 3rd. A phage display approach for rapid antibody humanization: Designed combinatorial V gene libraries. Proc. Natl. Acad. Sci. USA 1998, 95, 8910–8915. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Generic Name | Development Name, Trade Name or Drug Code | Target | Company | Format | Cancer Types Indicated | Highest Development Phase | Approved Year | Phage Display Technology | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Atezolizumab | Tecentriq | PD-L1 | Roche | IgG1k | UC, UBC, BC, NSCLC | NSCLC 2016/UC 2016/UBC 2017/BC 2019 | Genentech/HuCAL | [38,39] | |
| Moxetumomab Pasudotox-tdfk | Lumoxiti | CD22 | Medimmune/AstraZeneca | IgG1 | HCL | 2018 | CAT | [40,41,42] | |
| Necitumumab | IMC-11F8/Portrazza | EGFR | Lilly/AstraZeneca | igG1k | NSCLC | 2015 | Dyax | [43,44] | |
| Ramucirumab | Cyramza | VEGFR2 | Lilly/Imclone | IgG1 | GC, NSCLC, CC, HCC | GC, NSCLC 2014/CC 2015/HCC 2019 | Dyax | [45,46] | |
| Avelumab | Bavencio | PD-L1 | EMD Serono, Pfizer | IgG1λ | RCC, MCC, metastatic UC | MUC, MCC 2017/RCC 2019 | Dyax | [47] | |
| Trebananib | AMG 386 | Angiopoietin 1 and 2 | Amgen | IgG | ovarian, peritoneal, fallopian tube | Phase 3 | N/A | Dyax | [48] |
| Darleukin (L19-IL2) | N/A | Extra-domain B of fibronectin | Philogen | scFv-IL2 fusion | Melanoma | Phase 3 | N/A | “Pini” library | [31] |
| Ganitumab | AMG 479 | IGF-1R | Amgen | IgG1 | pancreatic, colorectal, breast, NSCLC | Phase 2 | N/A | Dyax | [49] |
| Cixutumumab | IMC-A12 | IGF-1R | ImClone Systems Inc. | IgG1λ | NSCLC, metastatic melanoma of the eye, liver | Phase 2 | N/A | Dyax | [50] |
| Seribantumab | MM-121 | ErbB3 | Merrimack Pharmaceuticals, partner with Sanofi | IgG2 | advanced ovarian, hormone-sensitive BC, NSCLC, and HER2 negative neoadjuvant BC | Phase 2 | N/A | Dyax | [51] |
| Mapatumumab | TRM-1, HGS-ETR1 | TRAIL-4 | Human Genome Sciences, Inc., a GSK company | IgG1 | NSCLC, NHL, liver, cervical | Phase 2 | N/A | CAT | [52] |
| Carlumab | CNTO 888 | MCP-1 (CCL-2) | J&J | IgG1k | prostate cancer (currently terminated) | Phase 2 | N/A | MorphoSys | [53] |
| Adecatumumab | MT201 | EpCAM | Amgen | IgG1 | CC | Phase 2 | N/A | Micromet | [54] |
| -- | AMG 780 | Angiopoietin | Amgen | IgG2 | advanced solid tumor | Phase 1 | N/A | Dyax | [55] |
| -- | IMC-3C5 | VEGFR-3 | ImClone | IgG1 | advanced solid tumor | Phase 1 | N/A | Dyax | [56] |
| Imalumab | Anti-MIF/BAX69 | MIF | Baxter | IgG1 | CC, advanced solid tumors | Phase 2 | N/A | Dyax | [57] |
| -- | MOR202 | CD38 | MorphoSys | IgG1 | MM | Phase 1/2 | N/A | MorphoSys | [58] |
| Anetumab ravatansine | BAY 94-9343 | Mesothelin | Bayer | IgG1λ, maytansinoid tubulin inhibitor DM4 conjugate | adenocarcinoma, mesothelioma, non-squamous NSCLC | Phase 2 | N/A | MorphoSys | [59] |
| Tarextumab | OMP-59R5 | Notch 2 and Notch 3 receptors | OncoMed | IgG2κ | solid tumors, small cell lung, pancreatic | Phase 1 | N/A | MorphoSys | [60] |
| Vantictumab | OMP-18R5 | Frizzled 7 receptor | OncoMed | IgG2λ | solid tumors | Phase 1 | N/A | MorphoSys | [61] |
| Samalizumab | ALXN6000 | CD200 | Alexion | IgG2 | B cell CLL, MM | Phase 1/2 | N/A | Alexion | [62] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Wang, Z. Monoclonal Antibody Development for Cancer Treatment Using the Phage Display Library Platform. Biologics 2024, 4, 55-74. https://doi.org/10.3390/biologics4010005
Zhang T, Wang Z. Monoclonal Antibody Development for Cancer Treatment Using the Phage Display Library Platform. Biologics. 2024; 4(1):55-74. https://doi.org/10.3390/biologics4010005
Chicago/Turabian StyleZhang, Tiantian, and Zhe Wang. 2024. "Monoclonal Antibody Development for Cancer Treatment Using the Phage Display Library Platform" Biologics 4, no. 1: 55-74. https://doi.org/10.3390/biologics4010005
APA StyleZhang, T., & Wang, Z. (2024). Monoclonal Antibody Development for Cancer Treatment Using the Phage Display Library Platform. Biologics, 4(1), 55-74. https://doi.org/10.3390/biologics4010005