



Biomolecules 2021, 11(11), 1647; https://doi.org/10.3390/biom11111647 - 8 Nov 2021
Cited by 2 | Viewed by 2164
Abstract
►
Show Figures
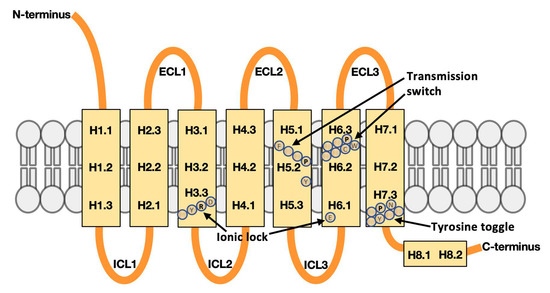
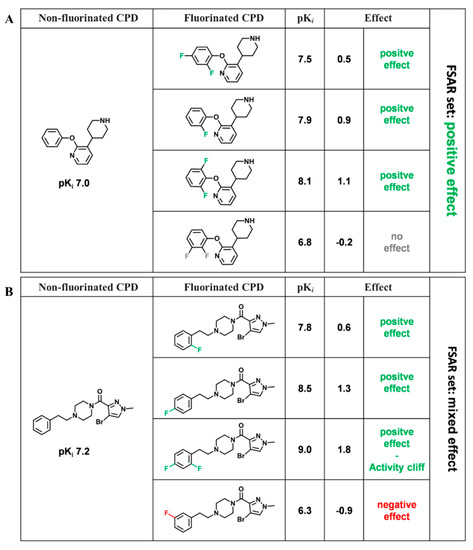
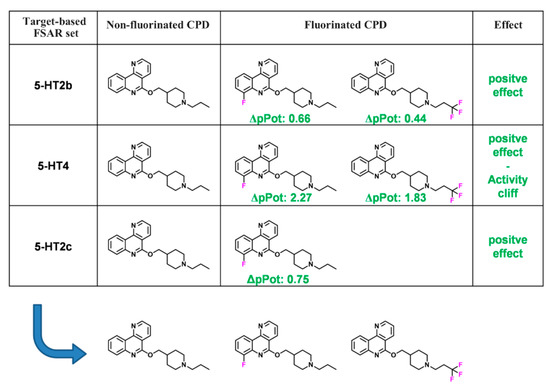
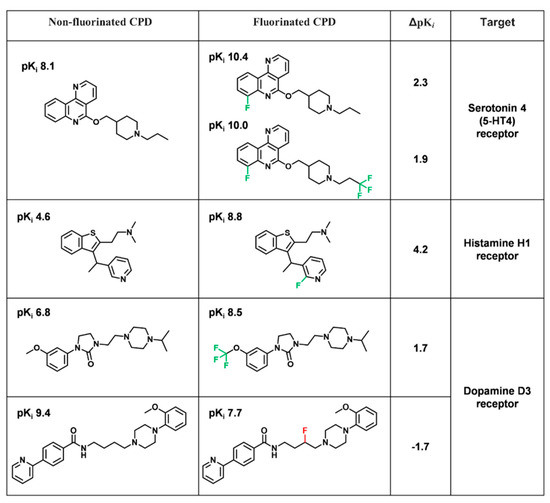
Currently, G protein-coupled receptors are the targets with the highest number of drugs in many therapeutic areas. Fluorination has become a common strategy in designing highly active biological compounds, as evidenced by the steadily increasing number of newly approved fluorine-containing drugs. Herein, we
[...] Read more.
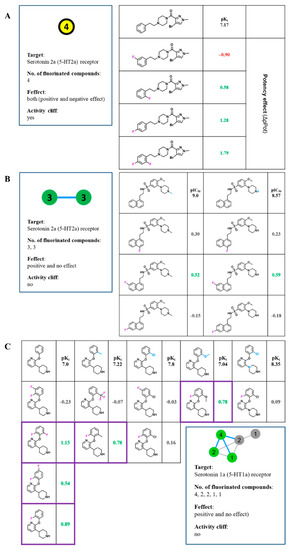
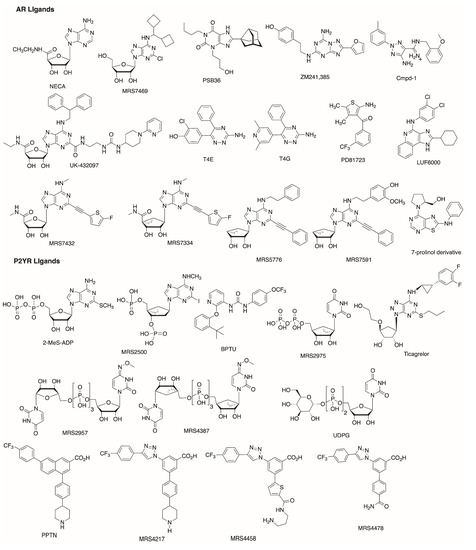
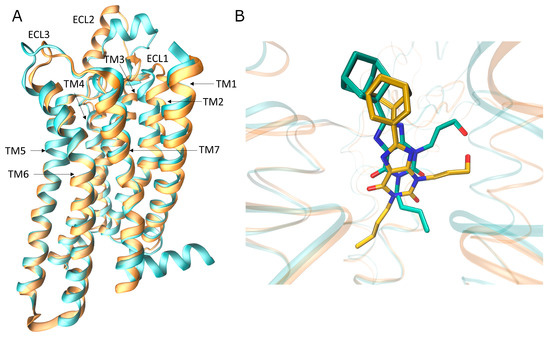
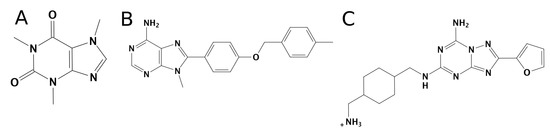
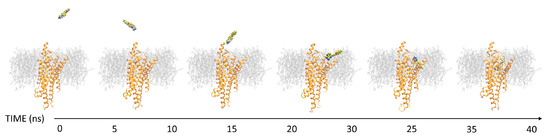
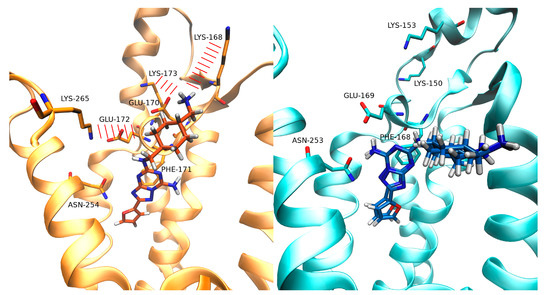
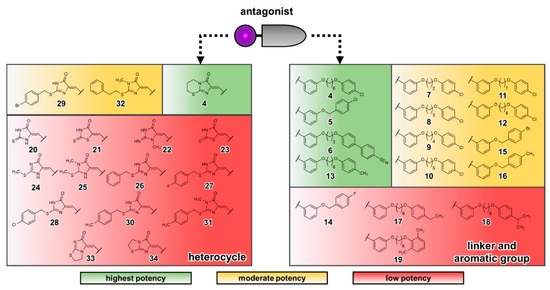
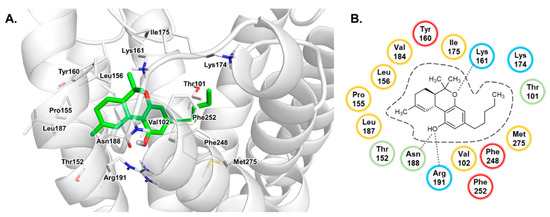
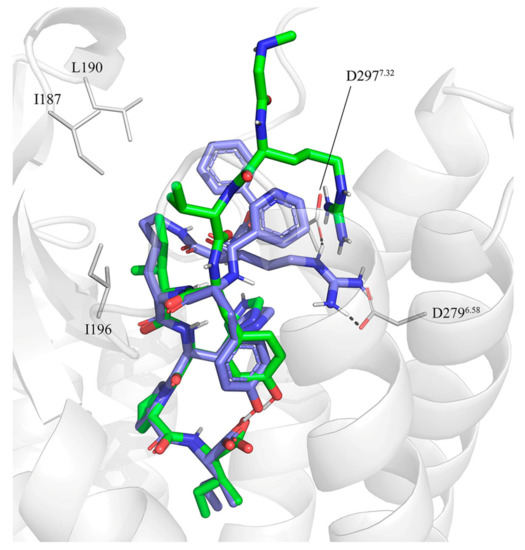
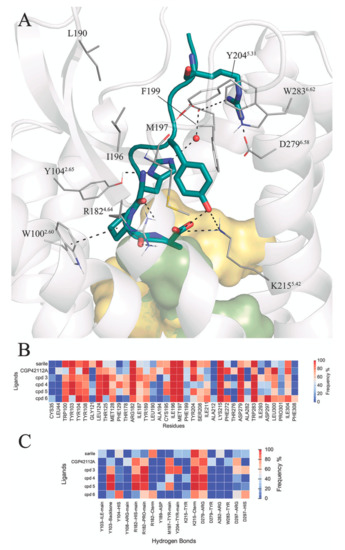
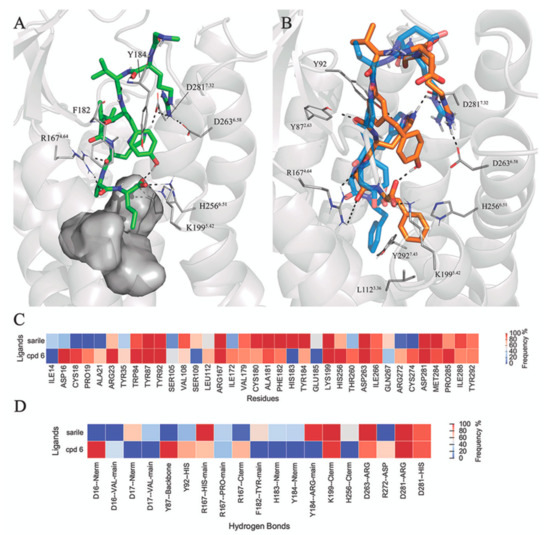
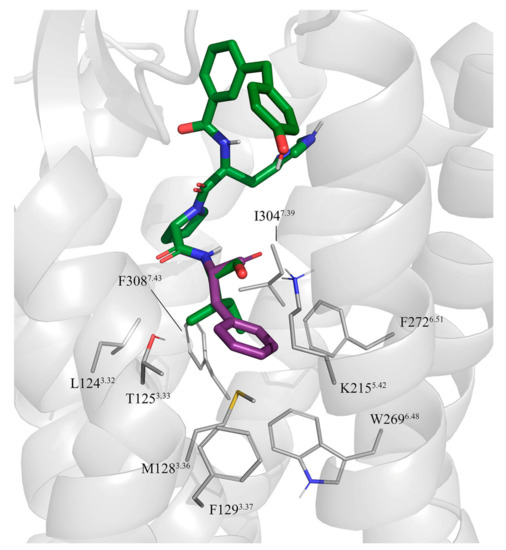
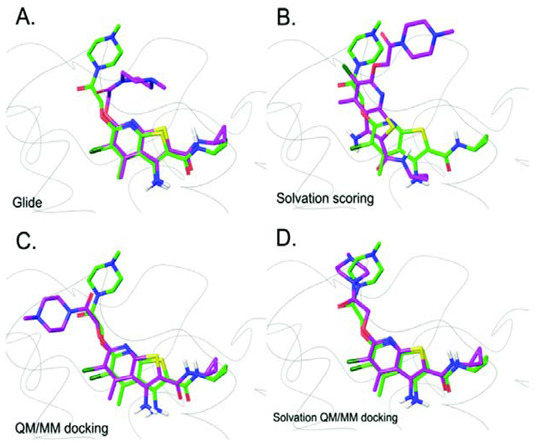
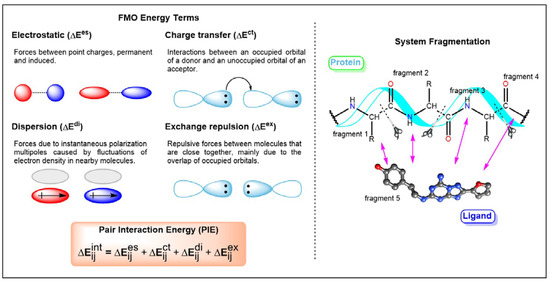
Currently, G protein-coupled receptors are the targets with the highest number of drugs in many therapeutic areas. Fluorination has become a common strategy in designing highly active biological compounds, as evidenced by the steadily increasing number of newly approved fluorine-containing drugs. Herein, we identified in the ChEMBL database and analysed 1554 target-based FSAR sets (non-fluorinated compounds and their fluorinated analogues) comprising 966 unique non-fluorinated and 2457 unique fluorinated compounds active against 33 different aminergic GPCRs. Although a relatively small number of activity cliffs (defined as a pair of structurally similar compounds showing significant differences of activity −ΔpPot > 1.7) was found in FSAR sets, it is clear that appropriately introduced fluorine can increase ligand potency more than 50-fold. The analysis of matched molecular pairs (MMPs) networks indicated that the fluorination of the aromatic ring showed no clear trend towards a positive or negative effect on affinity; however, a favourable site for a positive potency effect of fluorination was the ortho position. Fluorination of aliphatic fragments more often led to a decrease in biological activity. The results may constitute the rules of thumb for fluorination of aminergic receptor ligands and provide insights into the role of fluorine substitutions in medicinal chemistry.
Full article
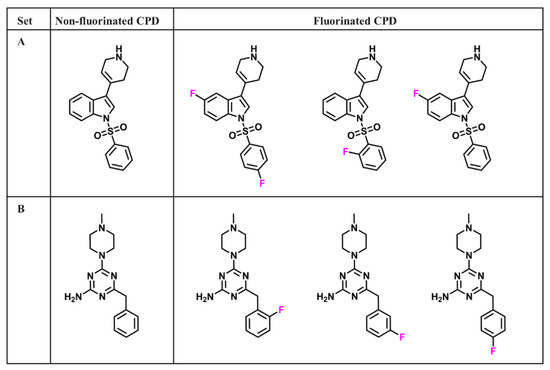
Figure 1
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}