1. Introduction
Photocatalytic water splitting is an attractive approach to addressing issues related to energy and the environment by converting renewable solar energy into chemical energy stored in the form of hydrogen [
1,
2]. However, this requires developing efficient semiconductor photocatalysts capable of using visible light, which accounts for close to 54% of solar energy to achieve sufficient solar-to-hydrogen energy conversion efficiencies with reasonable quantum efficiencies [
1]. Perovskite-type semiconducting oxynitrides with the general formula AB(N,O)
3 (where A and B represent relatively large twelve-coordinated cations, such as Ca
2+, Sr
2+, Ba
2+ and La
3+ and smaller six-coordinated cations, such as Ti
4+, Nb
5+ and Ta
5+, respectively) are promising visible-light-driven photocatalysts. These materials exhibit intense visible light absorption, tunable compositions and band structures, and stable crystal structures [
3,
4,
5,
6]. In particular, Nb-based perovskite-type oxynitrides have long absorption edge wavelengths (
λmax) [
6,
7]. Among these, LaNbN
2O exhibits an especially high
λmax of approximately 750 nm, [
7] corresponding to bandgap energy of 1.65 eV, that is superior to the values for CaNbNO
2 (
λmax = 600 nm), [
8] SrNbNO
2 (
λmax = 690 nm) [
9,
10] and BaNbNO
2 (
λmax = 740 nm), [
11,
12,
13] and so has been the subject of research and development efforts.
The oxygen evolution reaction is a half-reaction within the water splitting process, and many AB(N,O)
3 oxynitrides show decent activity in terms of promoting this reaction by photocatalysis [
5,
14,
15,
16]. Despite this, LaNbN
2O has rarely been applied to photocatalytic water oxidation. This is primarily because it is difficult to synthesize LaNbN
2O having suitable semiconducting characteristics. LaNbN
2O is typically obtained by heating oxides containing La and Nb under an NH
3 flow [
7,
17]. During this nitridation process, Nb
5+ ions are more readily reduced to Nb
4+, and Nb
3+ than are their Ta analogs, such that anion defects are generated [
6]. This leads to the formation of mid-gap defect states and the frequent formation of impurity phases, such as rock-salt-type Nb(N,O). Hence, it is necessary to apply moderate nitridation conditions, such as lowering the temperature and shortening the duration, to limit the reduction of Nb
5+. Unfortunately, these adjustments also produce an oxynitride with a low degree of crystallinity, increasing the extent of charge carrier recombination. Thus, it is necessary to investigate reduced nitridation times as an approach to producing LaNbN
2O with properties suitable for efficient photocatalytic O
2 evolution.
Recently, the engineering of precursor oxides based on tuning both the crystal structure and composition has been studied extensively to synthesize active oxynitride photocatalysts. Wang et al. prepared LaNbN
2O exhibiting a remarkable apparent quantum yield (AQY) of 0.82% at 420 nm during oxygen evolution from an aqueous AgNO
3 solution after being loaded with CoO
x as an oxygen evolution cocatalyst [
18]. This material was obtained by nitriding the layered oxide LaKNaNbO
5, which rapidly generated a plate-like LaNbN
2O shell with exposed (010) facets surrounding a LaKNaNbO
5 core. Both K and Na species were volatilized during the nitridation, and the matching of the La
3+ and Nb
5+ ion arrangements of the LaKNaNbO
5 and LaNbN
2O yielded highly crystalline LaNbN
2O having a low defect density. In other work, BaTaNO
2 that was highly active during the oxygen evolution reaction was prepared by nitriding a perovskite-type oxide having the nominal formula (Na
1/4Ba
3/4)(Zn
1/4 Ta
3/4)O
3 (in which the Ba/Ta ratio was unity), with the concurrent volatilization of Na and Zn [
15]. This material displayed a high AQY of 11.9% at 420 nm during the photocatalytic oxygen evolution reaction. The benefits of matching the arrangements of cationic components during the nitridation of oxides to oxynitrides have also been reported for other material systems [
19,
20,
21]. Hence, designing a stoichiometric and isostructural oxide precursor for conversion into an oxynitride photocatalyst has attracted significant attention as a possible means of enhancing the photocatalytic activity of Nb-based oxynitrides.
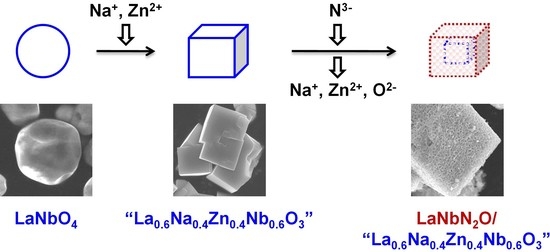
Based on the above, the present work designed a stoichiometric, isostructural precursor oxide having the nominal formula La0.6Na0.4Zn0.4Nb0.6O3 to prepare LaNbN2O via nitridation. The volatilization of Na and Zn species from this precursor was expected to promote the nitridation to a greater extent than using LaNbO4. The precursor oxide synthesized in this study was found to consist of a cuboid perovskite-type oxide resembling La0.5Na0.5Zn0.33Nb0.67O3, together with spherical LaNbO4 particles and several other impurities. Despite this lack of purity, when modified with a CoOx cocatalyst, the LaNbN2O-based material obtained from nitridation of this precursor exhibited more than five times greater photocatalytic evolution activity during the oxygen evolution reaction under visible light (with an AQY of 1.7% at 420 nm) as compared with LaNbN2O made by the nitridation of LaNbO4. These results demonstrate the possibility of using structurally modified precursor oxides to prepare oxynitride photocatalysts with enhanced activity based on reduced nitridation durations.
2. Results and Discussion
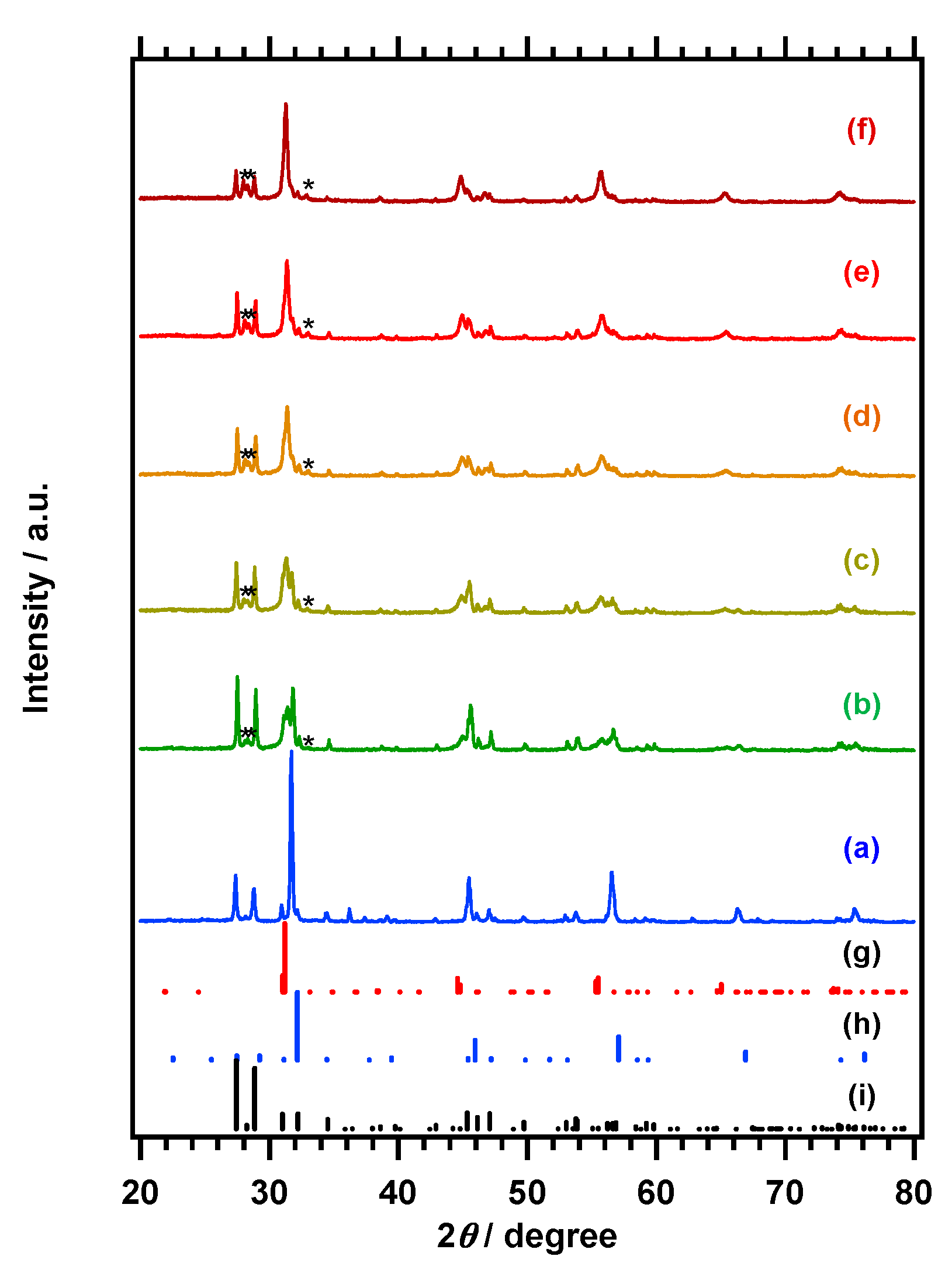
Figure 1A,B respectively presents the X-ray powder diffraction (XRD) patterns and diffuse-refrectance specopy (DRS) data obtained from the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 and LaNbO
4 and their products generated by nitridation at 1198 K for 2 h. Reference patterns for La
0.5Na
0.5Zn
0.33Nb
0.67O
3 (International Centre for Diffraction Data—powder diffraction file (ICDD-PDF): 00-043-0109), [
22] LaNbO
4 (ICDD-PDF: 01-071-1405), [
23] La
3NbO
7 (ICDD-PDF: 01-071-1345) [
24] and LaNbN
2O (ICDD-PDF: 01-078-7116) [
25] are also presented for comparison purposes in
Figure 1A. The LaNbO
4 sample was found to comprise a single-phase, and its pattern exhibits intense peaks at 27.4° and 28.9° that are attributable to the (−121) and (121) planes of LaNbO
4, respectively. The elemental analysis data obtained using inductively coupled plasma optical emission spectroscopy (ICP-OES), presented in
Table 1, indicate that the La/Nb molar ratio in the product was 1.0, consistent with the chemical formula of LaNbO
4. This material showed a light absorption edge at approximately 350 nm. The main XRD peaks produced by the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 resemble that of the perovskite-type compound La
0.5Na
0.5Zn
0.33Nb
0.67O
3, with a characteristic intense peak at 32.2° that is absent in the LaNbO
4 pattern. However, the peaks produced by the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 are positioned at lower diffraction angles compared to those in the La
0.5Na
0.5Zn
0.33Nb
0.67O
3 pattern. As shown in
Table 1, the La:Na:Zn:Nb molar ratio in the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 sample was 1.16:0.39:0.73:1.00, indicating a greater than 10% surplus and shortage, respectively, of La and Na in the material. The specimen also contained a slight excess of Zn. The XRD pattern and the light absorption onset at approximately 350 nm both suggest that the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 sample included a significant amount of LaNbO
4 as a byproduct. Moreover, the spectrum of the material contains an absorption shoulder tailing to approximately 400 nm that is characteristic of ZnO, even though this compound was not detected in the XRD pattern. Changing the calcination time did not have any significant effect on the XRD pattern of the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 synthesized by the flux method, and LaNbO
4 remained as a byproduct in each case. Therefore, it was impossible to determine the composition or crystal structure of the perovskite-type phase in the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 specimens. Nevertheless, we refer to this oxide herein as La
0.6Na
0.4Zn
0.4Nb
0.6O
3 based on the nominal molar ratio in the starting mixture for simplicity.
Scanning electron microscope (SEM) observations showed that the addition of Na and Zn precursors during the preparation of the oxide material significantly changed the particle morphology. The images obtained from the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 and LaNbO
4 and their products following nitridation at 1198 K for 2 h are presented in
Figure 2. The LaNbO
4 sample consisted of spherical particles with unclear edges and had particle sizes primarily in the range of 2–4 μm. In contrast, the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 was made of cuboid particles with clear surfaces and edges, 2–4 μm in size, in addition to spherical particles similar to those observed in the LaNbO
4 sample. The cuboid particles were confirmed to include Na and Zn by SEM-EDS and thus were attributed to a perovskite-type oxide similar to La
0.5Na
0.5Zn
0.33Nb
0.67O
3. A scanning transmission electron miscroscope (STEM) image of one such cuboid La
0.6Na
0.4Zn
0.4Nb
0.6O
3 particle is presented in
Figure 3 and shows clear lattice fringes over a wide range, indicating the single-crystalline nature of the particle. In fact, an selected area electron diffraction (SAED) pattern for one of these particles showed clear diffraction spots that provided further evidence for the single-crystalline nature of the material. Notably, when the heating duration to prepare the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 was shortened from 25 to 10 h, the product appeared to include more agglomerated cuboid particles. This presumably occurred because sufficient dissolution and recrystallization did not occur during the shorter heating time in the molten salt flux.
As shown in
Figure 1, the XRD pattern for the nitrided La
0.6Na
0.4Zn
0.4Nb
0.6O
3 contains a peak at approximately 31°, attributable to the (002) and (200) planes of LaNbN
2O [
25]. Some peaks related to the original La
0.6Na
0.4Zn
0.4Nb
0.6O
3 are also present, but their intensity is significantly weaker following nitridation, whereas the peaks ascribed to LaNbO
4 are essentially unchanged. Similar results were observed following the nitridation of LaNbO
4, in that peaks attributable to the LaNbN
2O phase were generated, but the LaNbO
4 peaks were barely weakened. These observations indicate the high reactivity of the perovskite-type oxide phase during the nitridation. Elemental analysis by ICP-OES established that La:Na:Zn:Nb molar ratio for the product obtained from nitridation of the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 at 1198 K for 2 h was 1.14:0.27:0.07:1.00. Therefore, the La component was preserved, and the Zn component was volatilized to a greater extent than the Na component during nitridation. It should also be noted that the La/Nb ratio was maintained during nitridation of LaNbO
4, and so the volatilization of La was evidently negligible. The excess of La species resulted in the formation of LaNbO
4 as a byproduct. La
3NbO
7 also accounted for a portion of the excess La species because small peaks attributable to La
3NbO
7 were observed after the nitridation. The remaining Na and Zn were presumably doped into the LaNbN
2O material, which may have affected the semiconducting properties of the nitridation product. The absorption edge wavelength as estimated from the onset of light absorption was approximately 700 nm. This wavelength was somewhat shorter than the value of 750 nm reported for LaNbN
2O and the light absorption onset observed for the nitridation product obtained from LaNbO
4 [
17,
18]. Notably, the nitridation product resulting from La
0.6Na
0.4Zn
0.4Nb
0.6O
3 exhibited a brighter color than that produced by LaNbO
4. It is suspected that the nitridation of LaNbO
4 into LaNbON
2 was too slow because it required significant rearrangement of the ions, such that anion vacancies and reduced Nb species were present in the product.
As shown in
Figure 2, the products resulting from nitridation of both La
0.6Na
0.4Zn
0.4Nb
0.6O
3 and LaNbO
4 exhibited roughened surfaces, although the sizes and contours of the original particles were fairly well preserved. These results are attributed to the volatilization of the constituent elements in La
0.6Na
0.4Zn
0.4Nb
0.6O
3 and to the increased density of the material obtained from LaNbO
4. Such effects are commonly observed following the nitridation of transition metal oxides [
12,
13,
16,
18,
19]. The structure of the nitridation product of La
0.6Na
0.4Zn
0.4Nb
0.6O
3 was closely inspected using STEM. As shown in
Figure 3e, the product particles had characteristic dense solid cores and porous shells, although the entire particles became porous when the particle size was sufficiently small. In contrast, the nitridation product generated from LaNbO
4 maintained the initial dense structure. Evidently, the formation of a porous structure due to the volatilization of the Na and Zn components effectively promoted nitridation to LaNbN
2O. However, it is arguable if Na and Zn species’ volatilization is a driving force to promote the nitridation because the composition of the starting oxide and the rates of Na and Zn evaporation and the exchange of O and N were not controlled.
Nb 3
d X-ray photoelectron spectroscopy (XPS) data were acquired to study the surface chemical states of the Nb species in these materials, and
Figure 4 provides the XPS spectra for La
0.6Na
0.4Zn
0.4Nb
0.6O
3 and LaNbO
4 and for their products following nitridation at 1198 K for 2 h. The Nb 3
d spectra of the oxide samples were deconvoluted to give a single doublet peak (representing Nb 3
d5/2 and 3
d3/2, with a peak separation of 2.75 eV) attributed to Nb
5+. In contrast, the Nb 3
d XPS spectra of the nitrided samples could be deconvoluted into two doublet peaks, and the Nb species associated with higher and lower binding energies are attributable to Nb
5+ and reduced Nb (Nb
4+ or Nb
3+) species, respectively. The Nb
5+ peaks produced by the nitrided samples were also positioned at lower binding energies than those generated by the oxide materials because the Nb
5+ ions formed bonds to less electronegative N
3− ions rather than to O
2− [
26]. The incorporation of N
3− in the material was confirmed based on the N 1
s peak at a binding energy of approximately 396 eV (
Figure S1). The proportions of reduced Nb species relative to the total Nb were almost the same for the samples obtained from nitridation of La
0.6Na
0.4Zn
0.4Nb
0.6O
3 and LaNbO
4. Therefore, using La
0.6Na
0.4Zn
0.4Nb
0.6O
3 did not have a unique effect in terms of suppressing the reduction of Nb
5+ during the nitridation.
Based on the above characterizations, it is clear that the addition of volatile Na and Zn components can modify the structure of LaNbO4 to provide a perovskite-type oxide and also promote nitridation to give a LaNbN2O phase although La0.6Na0.4Zn0.4Nb0.6O3 was not obtained as a pure material and the reduction of Nb5+ species during the nitridation was not suppressed. Subsequently, the effect of the nitridation conditions of La0.6Na0.4Zn0.4Nb0.6O3 on the properties of the products was investigated to gain additional insights concerning the nitridation process.
The XRD patterns and DRS spectra obtained from the products generated from La
0.6Na
0.4Zn
0.4Nb
0.6O
3 by nitridation at 1198 K for varying durations are presented in
Figure 5 and
Figure 6, respectively. Following the initial hour of nitridation, the diffraction peak associated with the perovskite-type oxide weakened significantly, while peaks attributed to the LaNbN
2O phase emerged, and the peaks related to LaNbO
4 also became more intense. In addition, peaks assigned to La
3NbO
7 became observable. As the nitridation time was prolonged, the diffraction peaks from the LaNbN
2O phase became more prominent and sharper, indicative of improved crystallinity, while those ascribed to the perovskite-type oxide were diminished. Both LaNbO
4 and La
3NbO
7 remained in the product after nitridation for 3 h. Elemental analysis by ICP-OES indicated that most Zn in La
0.6Na
0.4Zn
0.4Nb
0.6O
3 was vaporized during the initial hour of nitridation, whereas the evaporation of Na was slow, and approximately half of the initial quantity of Na remained after 3 h. This difference in the evaporation rates of Na
+ and Zn
2+ species would be expected to generate a La-rich byproduct (La
3NbO
7) in the initial stage of nitridation. In contrast, the change in the La amount was less than 3%. The nitridation product at 1 h exhibited an onset of light absorption at approximately 700 nm and continuous background absorption beyond the onset. As the heating duration was extended, the absorption onset remained largely unchanged, but the background absorption became stronger. These results suggest that the LaNbN
2O phase was produced during the very early stage of the nitridation process. Background absorption beyond the absorption edge wavelength is commonly associated with the presence of reduced Nb species and anion vacancies [
6,
7]. Similar trends were observed when the heating time was fixed at 2 h, and the nitridation temperature was varied (
Figures S2 and S3). The quantity of the perovskite-type oxide was reduced at higher temperatures, with the concurrent emergence of the LaNbN
2O phase, and byproducts, such as LaNbO
4 and La
3NbO
7 persisted even when the nitridation temperature was increased to 1248 K. The product showed a light absorption onset of approximately 700 nm and the background light absorption became pronounced at higher nitridation temperatures. The volatilization of Na and Zn components also proceeded, although the pace of the former was slower.
The oxygen evolution activity of the LaNbN
2O-based samples produced via nitridation of the La
0.6Na
0.4Zn
0.4Nb
0.6O
3 at 1198 K for 2 h was examined after loading a CoO
x cocatalyst. The cocatalyst-loading amount and loading temperature were initially optimized, as depicted in
Figures S4 and S5. The oxygen evolution rate was maximized to be 115–117 μmol h
−1 at a Co-loading of 0.5 wt % and loading temperature of 773 K. This CoO
x cocatalyst is thought to provide active sites for oxygen evolution and to facilitate the extraction of photoexcited holes from the photocatalyst [
16]. However, excessive loading can result in aggregation of the cocatalyst and shading of the photocatalyst from incident light, both of which can lower activity. The CoO
x-loading temperature window that resulted in the most effective improvement of the O
2 evolution activity was found to be quite narrow, presumably reflecting the tendency of Nb-based oxynitride photocatalysts to react with NH
3.
Figure 7 shows the time courses of gas evolution during the oxygen evolution reaction using the products obtained from La
0.6Na
0.4Zn
0.4Nb
0.6O
3 and LaNbO
4 after nitridation at 1198 K for 2 h. The nitridation product from La
0.6Na
0.4Zn
0.4Nb
0.6O
3 exhibited an oxygen evolution rate of 117 μmol h
−1, which was more than five times produced from LaNbO
4. The corresponding AQY value was 1.7% at 420 nm, which is twice the highest previously reported yield for LaNbN
2O synthesized via the nitridation of LaKNaNbO
5 [
18]. The gradual decrease in the O
2 evolution rate over time seen in these data is ascribed to the photodeposition of Ag, which blocked the surface of the photocatalyst and shielded it from the incident light. A small quantity of nitrogen (at most 10 μmol) was also found to be produced due to oxidation of the sample surface during the first hour of the reaction, but this nitrogen evolution dropped to almost zero in the following reaction. These phenomena are typically observed during photocatalytic water oxidation reactions using (oxy)nitrides [
5,
27]. The significant improvement in the oxygen evolution rate of the LaNbN
2O-based photocatalyst synthesized via nitriding La
0.6Na
0.4Zn
0.4Nb
0.6O
3 demonstrates the importance of designing a suitable precursor oxide for LaNbN
2O. The matching of the crystal structure allowed for the prompt conversion of the starting oxide into the oxynitride phase during the nitridation. As a result, the density of defects generated during the rearrangement of the constituent elements of LaNbN
2O could be suppressed. However, the extent of Nb
5+ reduction was not appreciably lowered. Moreover, the remaining Na and Zn may have been doped into the LaNbN
2O material and have affected the photocatalytic activity. Further investigation is needed to verify the origin of the activity enhancement of the present LaNbN
2O-based photocatalyst.
The photocatalytic oxygen evolution rates observed when using the nitridation products produced by La
0.6Na
0.4Zn
0.4Nb
0.6O
3 employing various conditions are presented in
Table 1. When using a fixed nitridation temperature of 1198 K, the rate of oxygen evolution increased with increases in the nitridation time from 1 to 2 h, presumably because of increases in the amount and degree of crystallinity of the LaNbN
2O phase. Interestingly, the oxygen evolution rate dropped drastically when the nitridation duration was extended to 3 h, even though the crystallinity of the LaNbN
2O phase was increased. This effect was likely associated with increases in the concentrations of reduced Nb species and anion vacancies, as is evident from the DRS data and Nb 3
d XPS spectra in
Figure 6 and
Figure S6, respectively. At a fixed nitridation duration of 2 h, the rate of oxygen evolution was increased markedly by increasing the nitridation temperature from 1173 to 1198 K but then decreased rapidly with the further elevation of the temperature, likely for similar reasons (see
Figures S3 and S6).
3. Methods
La0.6Na0.4Zn0.4Nb0.6O3 samples were synthesized by a flux method using Na2CO3 (2 mmol, Wako Pure Chemicals, 99.8%), La2O3 (3 mmol, Kanto Chemicals, 99.99%), ZnO (4 mmol, Kanto Chemicals, 99%) and Nb2O5 (3 mmol, High Purity Chemical Laboratory, 99.9%) as raw materials, with a NaCl (50 mmol, Wako, 99.5%) flux. The precursors were first combined and ground in an alumina mortar for 30 min before being transferred to alumina crucibles. The mixture was subsequently heated in air to 1373 K with a temperature ramp of 10 K min−1, held at that temperature for 25 h (unless otherwise noted), and then cooled at 10 K min−1. The product was washed with distilled water to remove the flux and then dried in a vacuum oven at 313 K overnight. This precursor oxide is denoted herein as La0.6Na0.4Zn0.4Nb0.6O3, based on the nominal molar ratios in the starting mixture, even though it was a combination of several oxides. For comparison purposes, LaNbO4 was also synthesized from La2O3 (3 mmol) and Nb2O5 (3 mmol) as precursors in a NaCl flux (50 mmol) following a similar procedure, but without using Na and Zn sources. The precursors were each calcined at 1373 K for 25 h.
The as-prepared La0.6Na0.4Zn0.4Nb0.6O3 was subjected to nitridation to produce LaNbN2O. In this process, a quantity (0.5 g) of the La0.6Na0.4Zn0.4Nb0.6O3 was transferred to an alumina tube and nitrided at 1173–1248 K for 1–3 h under a 200 mL min−1 flow of gaseous NH3. A quantity (0.5 g) of LaNbO4 was also nitrided in a similar manner, with heating at 1198 K for 2 h, to produce a control sample made without employing Na2CO3 or ZnO.
The CoO
x cocatalyst was deposited by impregnation followed by heating under a flow of NH
3 gas [
16]. During this impregnation, a quantity of the photocatalyst powder (0.3 g) was dispersed in a specific volume of an aqueous Co(NO
3)
2·6H
2O solution, followed by a 5 min sonication. The amount of Co added was 0.5 wt % concerning the mass of the photocatalyst powder. Subsequently, the material was completely dried by heating on a water bath at approximately 353 K, after which the powder was collected and heated at 773 K for 1 h in a 200 mL min
−1 flow of NH
3.
The photocatalyst samples were analyzed by XRD (MiniFlex 300, Rigaku (Tokyo, Japan); Cu Kα) over the 2θ range of 10° to 60°, and UV-visible DRS (V-670, JASCO, Nagano, Japan) data were acquired over the range of 200–1000 nm. The total reflectance was converted into Kubelka–Munk function (), where stands for the diffuse-reflectance measured concerning the standard. Spectralon was used as a standard white material. SEM images were acquired with a Phenom ProX desktop scanning electron microscope (Thermo Fisher Scientific, Waltham, MA, USA) and JSM-7600 F microscopes (JEOL, Tokyo, Japan). EDS data were acquired using a Phenom ProX (Thermo Fisher Scientific); STEM images were obtained with an HD2300A instrument (Hitachi High-Tech, Tokyo, Japan). High-resolution transmission electron microscopy (TEM) images and SAED patterns were acquired using a JEM-2010 (JEOL, Japan). XPS data were acquired with a PHI Quantera II spectrometer (ULVAC-PHI. INC, Chigasaki, Japan) incorporating an Al Kα X-ray source and the binding energy for the C 1s peak at 285.0 eV was used for calibration purposes. The elemental composition of each specimen was determined by ICP-OES (ICPS-8100, Shimadzu, Kyoto, Japan). Before analysis, the sample was transferred into a platinum crucible and heated with a molten salt mixture composed of K2CO3 and H3BO3. This material was then dissolved in a mixture of tartaric acid, hydrochloric acid and hydrogen peroxide and then diluted to a concentration suitable for analysis.
The photocatalytic reactions were carried out in a Pyrex top-irradiation type reaction vessel connected to a closed gas circulation system. In each trial, a quantity of the photocatalyst (0.3 g) loaded with the CoO
x cocatalyst was dispersed in 150 mL of a 50 mM aqueous AgNO
3 solution containing 0.20 g of La
2O
3, with the AgNO
3 and La
2O
3 serving as a sacrificial electron acceptor and a pH buffer, respectively [
27]. In this reaction, Ag
+ ions are reduced into metallic Ag by photoexcited electrons and water is oxidized into oxygen by photoexcited holes. Because of the deposition of Ag particles, the used photocatalyst cannot be reused. However, it is a common practice to use AgNO
3 in the photocatalytic material development stage, and developed materials can be utilized in overall water splitting reactions with improvement in the material synthesis and cocatalyst-loading technologies [
2,
5]. Successful examples include TaON and Ta
3N
5 [
20,
28]. Before photo-irradiation, the suspension in the reaction system was evacuated to completely remove air, after which Ar was introduced as a circulation gas. Following this, the suspension was irradiated from overhead using a 300 W xenon lamp equipped with a dichroic mirror and a cutoff filter (
λ ≥ 420 nm). The reactant solution was maintained at 288 K using a cooling water system during the reaction. The evolved gaseous products were analyzed using an integrated gas chromatography system consisting of a GC-8A chromatograph (Shimadzu, Japan) equipped with molecular sieve 5 Å columns and a thermal conductivity detector, with argon as the carrier gas.
The AQY for the oxygen evolution reaction was calculated as:
where
n(O
2) and
n(photons) are the number of oxygen molecules generated and the number of incident photons, respectively. The number of photons at 420 nm from a 300 W xenon lamp equipped with a bandpass filter was measured to be 3.9 × 10
20 photon h
−1 using an LS-100 spectroradiometer (EKO Instruments, Tokyo, Japan) by integrating the photon flux at different positions at the height of the suspension surface. The oxygen evolution was found to decrease over time due to the ongoing deposition of Ag, and so the AQY was calculated using
n(O
2) and
n(photons) values determined during the first hour of irradiation.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}