2. The Enthalpy-Entropy Interplay in Chiral Solutions
Spontaneous physical interactions in solution are, in general, associated with changes in free energy, ΔG, in the range of kcal/mol. The contributions of the change in enthalpy, ΔH, and the change due to entropy, TΔS, to the displayed change in free energy, are
a priori difficult to predict, but in general appear to be of the same order of magnitude, namely in the kcal/mol range. These basic thermodynamic parameters are readily determined and their scattered values for various protein-ligand interactions in water [
3], can illustrate their expected experimental range. However, translating the determined changes in enthalpy and entropy to the ensuing changes in the coordinates of the molecular assembly, is beyond the framework of classical thermodynamics. This can be achieved by employing specific technologies such as, for example, X-ray crystallography.
In the rather common processes of dilution or mixing of non reacting solutes in the same solvent, the changes in free energy are much smaller and expected to fall in the range of cal/mol, namely about 3 orders of magnitude below the range for specific interactions. Measurements of low energy processes, such as these, were neglected till recently, partially due to lack of interest but mostly because of shortage in suitable instruments. With the availability of accurate micro calorimetry set ups, in particular isothermal titration calorimetry (ITC) and related instrumentations [
4,
5], processes associated with changes in free energy in the range of cal/mol, or below, could be addressed. However, in such low energy processes, where no specific intermolecular interactions take place, resolution of the observed heat release or heat consumption, Q, to its thermodynamic components becomes non realistic. In this small energy region, the changes in enthalpy and entropy are actually intermingled and compensate for each other [
5]. In molecular terms they can be ascribed to the interplay between the overall change in order (entropy) and the parallel net distortion in the intermolecular bonds (enthalpy).
The observed heat release upon dilution of concentrated chiral solutions into water can be formally interpreted as the release of the excess chemical potential, µ
exc, which prevails in the concentrated solutions. This dilution process can be formulated as an increase of the activity coefficient γ (0 < γ < 1) of the solute towards 1:
Yet, in this particular somewhat trivial case, one can attribute the increase in γ to a decrease in “communication” between the solute molecules at distances greater than their net dimensions. This can be further ascribed to the dissociation of supra molecular solvent organizations which in the case of chiral solutions may imply solvent envelopes of a specific chiral configuration (see below). Thus, simple ITC dilution experiments of chiral solutions can lead to an estimated solvent configuration and its extension [
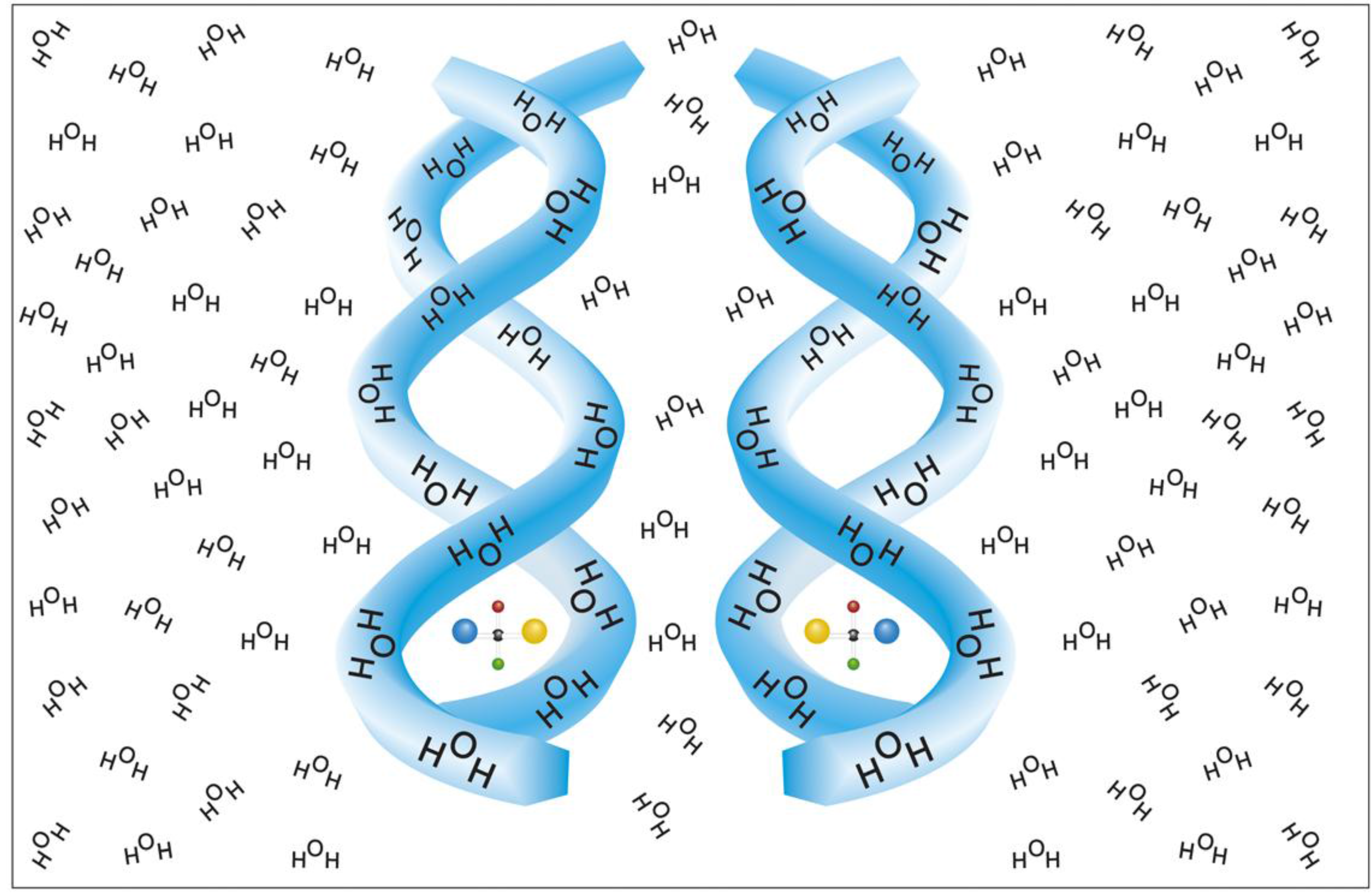
6]. The most plausible chiral configuration of a solvent envelope is that of a spiral, either right or left handed. As implied experimentally, such a putative spiral (or helix) can occupy a large number of solvent molecules and at high solute concentration can presumably form supra molecular chiral configurations, like double or triple helices, of solvent shells of adjacent solutes [
6]. A model for such chiral hydration envelopes, is presented in
Figure 1.
Figure 1.
A helical model for chiral hydration envelopes surrounding opposite enantiomers in an isotropic water environment [
6].
Figure 1.
A helical model for chiral hydration envelopes surrounding opposite enantiomers in an isotropic water environment [
6].
Homogeneous solutions of chiral substances are characterized by rotation of the optical axis of polarized light, a unique property which was already implemented by the great Louis Pasteur in 1848 [
7]. Thus, despite being fully isotropic, such solutions retain an overall asymmetry which can be readily evaluated by simple spectral tests. The quantitative parameter which corresponds to the degree of order which prevails in chiral solutions is the angle of rotation, α, of linearly polarized light recorded upon passage through the solution. It is linear with respect to concentration and optical path and could thus provide a quantitative scale for the overall asymmetry which prevails in the solution. This unique feature can provide an experimental quantitative bridging between entropy and order in chiral solutions [
1]. Furthermore, correlation between optical rotation and entropy can be extended to information capacity, expressed in energy units [
1], yet unprecedented achievement.
The recorded α in the common case of a homogeneous solution of a chiral substance, termed here as “chiral solution”, is the resultant of two levels of chiral asymmetries. The first is the overall asymmetry of the homogeneously distributed molecules of the chiral solute itself which, in principle, can be roughly evaluated by the optical activity of the same concentration in the vapor phase [
8,
9]. Its contribution to the displayed α is considerably smaller than that contributed by the chiral organization of the solvent molecules around the chiral centers of the solute [
1,
6,
9]. At high solute concentration such chiral envelopes can aggregate to supra molecular chiral assemblies which are dissociated upon dilution [
6]. Heat release, in the range of several cal/mol, ensuing such a dilution, was recently recorded in D and L alanine solutions and could be attributed to TΔS stored in the supra molecular chiral configuration of the intermolecular conjunctions of the chiral envelopes surrounding the individual chiral solutes [
6]. The negative entropy and its corresponding thermal energy, TΔS, stored in the individual chiral configuration of solvent envelopes, in addition to the solute overall asymmetry, is readily achieved by intermolecular racemization upon mixing [
1]. Specific values obtained by this procedure are presented in
Table 1.
3. Entropy and Order
In assemblies of macroscopic objects deviations from homogeneous distribution can be qualitatively presented in terms of “increased order”. Quantitative scales for the acquired “degree of order”, W, in such cases, can be inferred from statistical algorithms, initiated by Shannon and his associates and later by Brillouin [
10,
11]. It is of fundamental importance that under such statistical evaluation, W apparently correlates inversely with a term analogous to entropy, S, in statistical thermodynamics. In a homogeneous chiral solution the situation is quite unique. Despite being isotropic it stores an inherent degree of order expressed in reduction of entropy, S, which can be presented in terms of “negative entropy” or “configurational entropy”. The correlation between the degree of order, W, and the entropy in such systems is analogous to the macroscopic anisotropy [
10,
11] and is fundamentally formulated in the well-known Boltzmann equation:
where R is the gas constant.
At a complete hypothetical isotropy, where the system reaches its maximal degree of homogeneity or minimal order of W
0, the corresponding maximal entropy, S
0 provides the base line for evaluation of corresponding “negative entropy” ΔS stored in the homogeneous chiral solution:
The term TΔS represents the chemical potential stored in the ordered system which is released as thermal energy upon annulment.
6. Experimental Verification
A series of indirect evidence strongly suggest that the hydration layer surrounding chiral solutes, like amino acids, acquires a chiral configuration induced by their chiral centers [
1,
6]. The energy invested in the induction of the chiral twist in the hydration layer can predominantly be attributed to TΔS, the energy associated with “configurational entropy” [
2,
6]. This entropic energy amounts to only several cal/mol, about a thousandth of the enthalpy associated with the prevailing intermolecular interactions, like the hydrogen bonds. Thermal energies of this scale can be accurately monitored by microcalorimetry [
3,
4,
5].
TΔS of configurational entropy in chiral solution can be determined by heat release of two independent processes; heat of dilution and heat of intermaolecular racemization. The observed heat release upon dilution of concentrated chiral solutions into water can be formally interpreted as the release of the excess chemical potential, µ
exc, which prevails in the concentrated solutions. This dilution process can be formulated as an increase of the activity coefficient γ (0 < γ < 1) of the solute towards 1, formulated in Equation 1. In molecular terms, the solute molecules in such “ideal” solutions are separated by the bulk solvent beyond the level that permits static or dynamic correspondence between neighboring hydration layers. In line with this approach, all water molecules in the bulk are at a state which responds to the ordered hydration layers by adopting an overall ordered network which deviates from the isotropy prevailing in pure water. Furthermore, this putative overall degree of order increases with the solute concentration [
1,
6]. The hydration assemblies of the individual solutes at high concentrations presumably integrate to combined intermolecular assemblies which are tighter and more ordered than the hydration layer in dilute solutions. It has been thus shown, that heat release upon dilution of concentrated D or L-alanine in water has a TΔS component attributed to the disintegration of supramolecular chiral hydration assemblies [
6].



{kind=link}