
A Novel Sequence-Based Feature for the Identification of DNA-Binding Sites in Proteins Using Jensen–Shannon Divergence


Abstract
:1. Introduction
2. Results
2.1. Random Forest Classifier
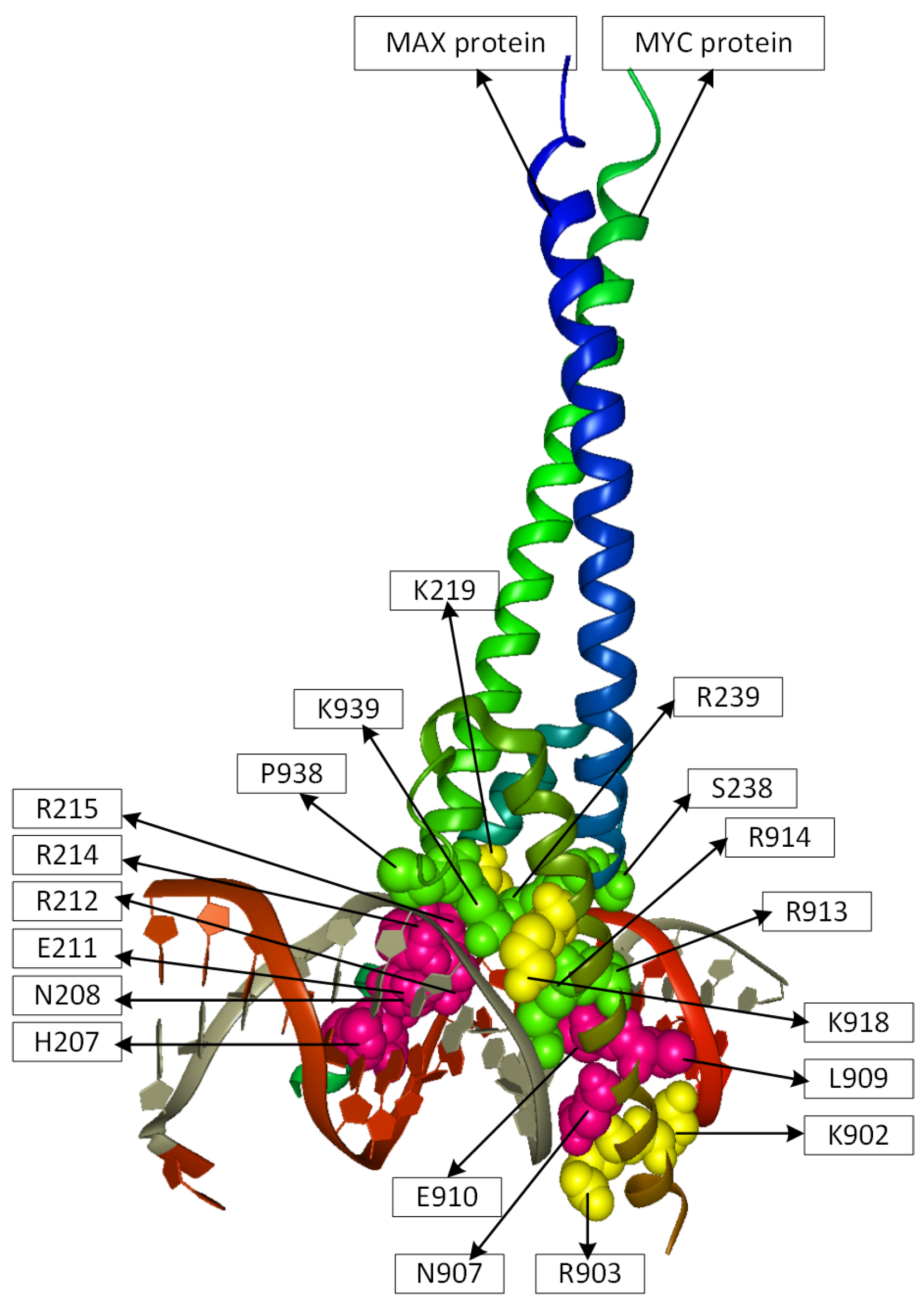
2.2. Position Analysis of the MYC-MAX Protein
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
Appendix A.1. Performance Measures with Standard Error

{kind=link}
| Feature | Sensitivity ± SE(%) | Specificity ± SE(%) | MCC ± SE(%) |
|---|---|---|---|
| 29.2 ± 2.20 | 96.3 ± 0.46 | 30.7 ± 0.95 | |
| + | 38.5 ± 3.04 | 94.9 ± 0.57 | 34.9 ± 1.7 |
| + | 41.0 ± 3.23 | 93.9 ± 0.57 | 35.0 ± 1.85 |
| + + | 41.4 ± 3.42 | 94.0 ± 0.51 | 34.8 ± 2.07 |
| + | 33.9 ± 2.32 | 95.8 ± 0.37 | 33.4 ± 1.36 |
| + + | 41.6 ± 3.05 | 95.0 ± 0.46 | 37.8 ± 2.19 |
| + + | 44.1 ± 3.12 | 94.0 ± 0.43 | 37.2 ± 2.37 |
| + + + | 43.9 ± 3.14 | 94.0 ± 0.40 | 37.0 ± 2.25 |
| + + | 36.7 ± 2.07 | 96.8 ± 0.27 | 39.8 ± 1.58 |
| + + + | 42.2 ± 2.70 | 95.8 ± 0.42 | 40.9 ± 1.95 |
| + + + | 44.7 ± 3.05 | 95.0 ± 0.38 | 40.3 ± 1.98 |
| + + + + | 44.4 ± 3.12 | 94.7 ± 0.39 | 39.3 ± 2.02 |
| Feature | Sensitivity ± SE(%) | Specificity ± SE(%) | MCC ± SE(%) |
|---|---|---|---|
| 28.6 ± 2.56 | 96.6 ± 0.47 | 35.0 ± 1.43 5 | |
| + | 39.5 ± 2.89 | 95.0 ± 0.55 | 40.7 ± 1.99 |
| + | 41.8 ± 3.02 | 94.3 ± 0.62 | 41.1 ± 2.05 |
| + + | 42.6 ± 3.25 | 94.2 ± 0.54 | 41.4 ± 2.37 |
| + | 33.4 ± 2.34 | 96.3 ± 0.38 | 38.6 ± 1.90 |
| + + | 42.4 ± 2.97 | 95.1 ± 0.61 | 43.6 ± 2.43 |
| + + | 44.8 ± 2.99 | 94.4 ± 0.56 | 43.8 ± 2.45 |
| + + + | 44.5 ± 3.04 | 94.4 ± 0.50 | 43.4 ± 2.35 |
| + + | 33.7 ± 2.48 | 97.5 ± 0.35 | 43.1 ± 2.05 |
| + + + | 41.9 ± 2.89 | 95.8 ± 0.55 | 45.0 ± 2.39 |
| + + + | 43.9 ± 2.89 | 95.2 ± 0.48 | 45.3 ± 2.32 |
| + + + + | 44.2 ± 2.91 | 94.9 ± 0.54 | 44.5 ± 2.24 |
Appendix A.2. RBscore Dataset Analysis
| Feature | Sensitivity | Specificity | MCC | AUC-ROC | AUC-PR |
|---|---|---|---|---|---|
| 0.458 | 0.974 | 0.476 | 0.866 | 0.460 | |
| + | 0.56 | 0.965 | 0.514 | 0.894 | 0.518 |
| + | 0.597 | 0.957 | 0.511 | 0.899 | 0.523 |
| + + | 0.591 | 0.958 | 0.511 | 0.90 | 0.526 |
| + | 0.512 | 0.97 | 0.501 | 0.878 | 0.476 |
| + + | 0.581 | 0.96 | 0.511 | 0.899 | 0.520 |
| + + | 0.611 | 0.953 | 0.508 | 0.903 | 0.526 |
| + + + | 0.613 | 0.953 | 0.509 | 0.902 | 0.528 |
| + + | 0.517 | 0.976 | 0.534 | 0.896 | 0.528 |
| + + + | 0.58 | 0.967 | 0.54 | 0.907 | 0.543 |
| + + + | 0.612 | 0.963 | 0.546 | 0.910 | 0.551 |
| + + + + | 0.601 | 0.962 | 0.531 | 0.909 | 0.546 |
| Feature | Sensitivity | Specificity | MCC | AUC-ROC | AUC-PR |
|---|---|---|---|---|---|
| 0.445 | 0.977 | 0.528 | 0.873 | 0.589 | |
| + | 0.553 | 0.968 | 0.579 | 0.899 | 0.643 |
| + | 0.57 | 0.962 | 0.572 | 0.900 | 0.642 |
| + + | 0.569 | 0.963 | 0.574 | 0.895 | 0.642 |
| + | 0.49 | 0.973 | 0.547 | 0.880 | 0.602 |
| + + | 0.578 | 0.963 | 0.583 | 0.902 | 0.648 |
| + + | 0.605 | 0.958 | 0.587 | 0.904 | 0.652 |
| + + + | 0.603 | 0.959 | 0.587 | 0.902 | 0.653 |
| + + | 0.499 | 0.98 | 0.584 | 0.895 | 0.641 |
| + + + | 0.57 | 0.968 | 0.595 | 0.908 | 0.661 |
| + + + | 0.592 | 0.965 | 0.60 | 0.908 | 0.665 |
| + + + + | 0.594 | 0.964 | 0.597 | 0.907 | 0.663 |
Appendix A.3. PreDNA Dataset Analysis
| Feature | Sensitivity | Specificity | MCC | AUC-ROC | AUC-PR |
|---|---|---|---|---|---|
| 0.378 | 0.977 | 0.41 | 0.840 | 0.391 | |
| + | 0.498 | 0.963 | 0.448 | 0.865 | 0.453 |
| + | 0.543 | 0.953 | 0.445 | 0.869 | 0.451 |
| + + | 0.538 | 0.956 | 0.453 | 0.869 | 0.455 |
| + | 0.393 | 0.975 | 0.417 | 0.847 | 0.402 |
| + + | 0.501 | 0.966 | 0.461 | 0.872 | 0.463 |
| + + | 0.545 | 0.959 | 0.465 | 0.876 | 0.468 |
| + + + | 0.523 | 0.958 | 0.449 | 0.875 | 0.465 |
| + + | 0.428 | 0.977 | 0.458 | 0.867 | 0.451 |
| + + + | 0.511 | 0.97 | 0.488 | 0.885 | 0.488 |
| + + + | 0.539 | 0.962 | 0.475 | 0.888 | 0.488 |
| + + + + | 0.539 | 0.961 | 0.47 | 0.886 | 0.488 |
| Feature | Sensitivity | Specificity | MCC | AUC-ROC | AUC-PR |
|---|---|---|---|---|---|
| 0.373 | 0.979 | 0.463 | 0.833 | 0.496 | |
| + | 0.485 | 0.962 | 0.495 | 0.858 | 0.540 |
| + | 0.496 | 0.953 | 0.475 | 0.858 | 0.534 |
| + + | 0.495 | 0.955 | 0.479 | 0.857 | 0.535 |
| + | 0.389 | 0.977 | 0.47 | 0.839 | 0.501 |
| + + | 0.49 | 0.963 | 0.501 | 0.863 | 0.550 |
| + + | 0.503 | 0.957 | 0.492 | 0.865 | 0.547 |
| + + + | 0.504 | 0.958 | 0.497 | 0.865 | 0.550 |
| + + | 0.395 | 0.98 | 0.488 | 0.858 | 0.530 |
| + + + | 0.48 | 0.968 | 0.511 | 0.874 | 0.563 |
| + + + | 0.506 | 0.962 | 0.51 | 0.873 | 0.560 |
| + + + + | 0.499 | 0.96 | 0.498 | 0.871 | 0.555 |
References
- Liu, B.; Wang, S.; Wang, X. DNA binding protein identification by combining pseudo amino acid composition and profile-based protein representation. Sci. Rep. 2015, 5, 15479. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Westhof, E. Prediction of nucleic acid binding probability in proteins: A neighboring residue network based score. Nucleic Acids Res. 2015, 43, 5340–5351. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Zhang, Z.; Lin, B.; Schroeder, M.; Huang, B. MetaDBSite: A meta approach to improve protein DNA-binding sites prediction. BMC Syst. Biol. 2011, 5 (Suppl. S1), S7. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Guo, J.; Liu, H.D.; Xie, J.M.; Sun, X. Sequence-based prediction of DNA-binding residues in proteins with conservation and correlation information. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, H.; Duan, X.; Ding, Y.; Wu, H.; Bai, Y.; Sun, X. Prediction of DNA-binding residues in proteins from amino acid sequences using a random forest model with a hybrid feature. Bioinformatics 2009, 25, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xu, J.; Fan, S.; Xu, R.; Zhou, J.; Wang, X. PseDNA-Pro: DNA-Binding Protein Identification by Combining Chou’s PseAAC and Physicochemical Distance Transformation. Mol. Inform. 2015, 34, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhou, J.; Wang, H.; He, Y.; Wang, X.; Liu, B. Identifying DNA-binding proteins by combining support vector machine and PSSM distance transformation. BMC Syst. Biol. 2015, 9 (Suppl. S1), S10. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Wang, S.; Wang, K.; Liu, X.; Liu, B. Identification of DNA-binding proteins by auto-cross covariance transformation. In Proceedings of the 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Washington, DC, USA, 9–12 November 2015; pp. 470–475.
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2016, in press. [Google Scholar] [CrossRef]
- Waris, M.; Ahmad, K.; Kabir, M.; Hayat, M. Identification of DNA binding proteins using evolutionary profiles position specific scoring matrix. Neurocomputing 2016, 199, 154–162. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, R.; He, Y.; Lu, Q.; Wang, H.; Kong, B. PDNAsite: Identification of DNA-binding Site from Protein Sequence by Incorporating Spatial and Sequence Context. Sci. Rep. 2016, 6, 27653. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Shanahan, H.P.; Berman, H.M.; Thornton, J.M. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003, 31, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Gromiha, M.M.; Sarai, A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics 2004, 20, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Langlois, R.E.; Zhao, G.; Lu, H. Structure based prediction of binding residues on DNA-binding proteins. In Proceedings of the IEEE 27th Annual International Conference of the Engineering in Medicine and Biology Society (IEEE-EMBS 2005), Shanghai, China, 1–4 September 2005; pp. 2611–2614.
- Ahmad, S.; Sarai, A. PSSM-based prediction of DNA binding sites in proteins. BMC Bioinform. 2005, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, I.B.; Gou, Z.; Li, R.; Hwang, S. Using evolutionary and structural information to predict DNA-binding sites on DNA-binding proteins. Proteins 2006, 64, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, S.J. Prediction of DNA-binding residues from sequence features. J. Bioinform. Comput. Biol. 2006, 4, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, S.J. BindN: A web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res. 2006, 34, W243–W248. [Google Scholar] [CrossRef] [PubMed]
- Ofran, Y.; Mysore, V.; Rost, B. Prediction of DNA-binding residues from sequence. Bioinformatics 2007, 23, i347–i353. [Google Scholar] [CrossRef] [PubMed]
- Siggers, T.W.; Honig, B. Structure-based prediction of C2H2 zinc-finger binding specificity: Sensitivity to docking geometry. Nucleic Acids Res. 2007, 35, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Tjong, H.; Zhou, H.X. DISPLAR: An accurate method for predicting DNA-binding sites on protein surfaces. Nucleic Acids Res. 2007, 35, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Nimrod, G.; Schushan, M.; Szilágyi, A.; Leslie, C.; Ben-Tal, N. iDBPs: A web server for the identification of DNA binding proteins. Bioinformatics 2010, 26, 692–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, C.; Yang, M.Q.; Yang, J.Y. BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst. Biol. 2010, 4 (Suppl. S1), S3. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Westhof, E. A Large-Scale Assessment of Nucleic Acids Binding Site Prediction Programs. PLoS Comput. Biol. 2015, 11, e1004639. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Friedrich, S.; Kurgan, L. A comprehensive comparative review of sequence-based predictors of DNA-and RNA-binding residues. Brief. Bioinform. 2015, 17, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Terribilini, M.; Wu, F.; Jernigan, R.L.; Dobbs, D.; Honavar, V. Predicting DNA-binding sites of proteins from amino acid sequence. BMC Bioinform. 2006, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Gou, Z.; Kuznetsov, I.B. DP-Bind: A web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics 2007, 23, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Huang, C.C.; Liu, Y.C.; Oyang, Y.J.; Huang, C.K. DNA-binding residues and binding mode prediction with binding-mechanism concerned models. BMC Genom. 2009, 10 (Suppl. S3), S23. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.C.; Li, Y.; Peng, C.; Moses, A.M.; Zhang, Z. Computational learning on specificity-determining residue-nucleotide interactions. Nucleic Acids Res. 2015, 43, 10180–10189. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, M.Q.; Yang, J.Y. Prediction of DNA-binding residues from protein sequence information using random forests. BMC Genom. 2009, 10 (Suppl. S1), S1. [Google Scholar] [CrossRef] [PubMed]
- Eggeling, R.; Roos, T.; Myllymäki, P.; Grosse, I. Inferring intra-motif dependencies of DNA binding sites from ChIP-seq data. BMC Bioinform. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Gültas, M.; Düzgün, G.; Herzog, S.; Jäger, S.J.; Meckbach, C.; Wingender, E.; Waack, S. Quantum coupled mutation finder: Predicting functionally or structurally important sites in proteins using quantum Jensen–Shannon divergence and CUDA programming. BMC Bioinform. 2014, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Mayer, C.E.; Söding, J. Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics 2008, 24, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Grosse, I.; Bernaola-Galván, P.; Carpena, P.; Román-Roldán, R.; Oliver, J.; Stanley, H.E. Analysis of symbolic sequences using the Jensen–Shannon divergence. Phys. Rev. E 2002, 65, 041905. [Google Scholar] [CrossRef] [PubMed]
- Gültas, M.; Haubrock, M.; Tüysüz, N.; Waack, S. Coupled mutation finder: A new entropy-based method quantifying phylogenetic noise for the detection of compensatory mutations. BMC Bioinform. 2012, 13, 225. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, Q.Z.; Liu, S.; Fan, G.L.; Zuo, Y.C.; Peng, Y. PreDNA: Accurate prediction of DNA-binding sites in proteins by integrating sequence and geometric structure information. Bioinformatics 2013, 29, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Krall, A.; Brunn, J.; Kankanala, S.; Peters, M.H. A simple contact mapping algorithm for identifying potential peptide mimetics in protein–protein interaction partners. Proteins 2014, 82, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA: Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, V.; Sommer, H.J.; Bruzda, W.; Zyczkowski, K. Random bistochastic matrices. J. Phys. A Math. Theor. 2009, 42, 36. [Google Scholar] [CrossRef]
- Bartlett, G.J.; Porter, C.T.; Borkakoti, N.; Thornton, J.M. Analysis of catalytic residues in enzyme active sites. J. Mol. Biol. 2002, 324, 105–121. [Google Scholar] [CrossRef]
- Panchenko, A.R.; Kondrashov, F.; Bryant, S. Prediction of functional sites by analysis of sequence and structure conservation. Protein Sci. 2004, 13, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.O.; Busch, M.; Kück, F.; Porfenenko, M.; Merkl, R. CLIPS-1D: Analysis of multiple sequence alignments to deduce for residue-positions a role in catalysis, ligand-binding, or protein structure. BMC Bioinform. 2012, 13, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Breiman, L. Bagging predictors. Mach. Learn. 1996, 24, 123–140. [Google Scholar] [CrossRef]

| Feature | Sensitivity | Specificity | MCC | AUC-ROC | AUC-PR |
|---|---|---|---|---|---|
| 0.292 | 0.963 | 0.307 | 0.777 | 0.313 | |
| + | 0.385 | 0.949 | 0.349 | 0.795 | 0.369 |
| + | 0.41 | 0.939 | 0.35 | 0.802 | 0.377 |
| + + | 0.414 | 0.94 | 0.348 | 0.800 | 0.376 |
| + | 0.339 | 0.958 | 0.334 | 0.794 | 0.338 |
| + + | 0.416 | 0.95 | 0.378 | 0.808 | 0.390 |
| + + | 0.441 | 0.94 | 0.372 | 0.817 | 0.401 |
| + + + | 0.439 | 0.94 | 0.37 | 0.814 | 0.399 |
| + + | 0.367 | 0.968 | 0.398 | 0.838 | 0.413 |
| + + + | 0.422 | 0.958 | 0.409 | 0.837 | 0.425 |
| + + + | 0.447 | 0.95 | 0.403 | 0.841 | 0.431 |
| + + + + | 0.444 | 0.947 | 0.393 | 0.835 | 0.423 |
| Feature | Sensitivity | Specificity | MCC | AUC-ROC | AUC-PR |
|---|---|---|---|---|---|
| 0.286 | 0.966 | 0.350 | 0.778 | 0.425 | |
| + | 0.395 | 0.95 | 0.407 | 0.801 | 0.487 |
| + | 0.418 | 0.943 | 0.411 | 0.807 | 0.494 |
| + + | 0.426 | 0.942 | 0.414 | 0.807 | 0.497 |
| + | 0.334 | 0.963 | 0.386 | 0.796 | 0.455 |
| + + | 0.424 | 0.951 | 0.436 | 0.814 | 0.513 |
| + + | 0.448 | 0.944 | 0.438 | 0.820 | 0.520 |
| + + + | 0.445 | 0.944 | 0.434 | 0.819 | 0.521 |
| + + | 0.337 | 0.975 | 0.431 | 0.830 | 0.517 |
| + + + | 0.419 | 0.958 | 0.450 | 0.832 | 0.535 |
| + + + | 0.439 | 0.952 | 0.453 | 0.836 | 0.539 |
| + + + + | 0.442 | 0.949 | 0.445 | 0.832 | 0.535 |
| Cut-Off | Feature | Sensitivity | Specificity | MCC | AUC-ROC | AUC-PR |
|---|---|---|---|---|---|---|
| 3.5 Å | + + | 0.517 | 0.976 | 0.534 | 0.896 | 0.528 |
| + + + | 0.58 | 0.967 | 0.54 | 0.907 | 0.543 | |
| + + + | 0.612 | 0.963 | 0.546 | 0.910 | 0.551 | |
| + + + + | 0.601 | 0.962 | 0.531 | 0.909 | 0.546 | |
| 5.0 Å | + + | 0.499 | 0.98 | 0.584 | 0.895 | 0.641 |
| + + + | 0.57 | 0.968 | 0.595 | 0.908 | 0.661 | |
| + + + | 0.592 | 0.965 | 0.60 | 0.908 | 0.665 | |
| + + + + | 0.594 | 0.964 | 0.597 | 0.907 | 0.663 |
| Cut-Off | Feature | Sensitivity | Specificity | MCC | AUC-ROC | AUC-PR |
|---|---|---|---|---|---|---|
| 3.5 Å | + + | 0.428 | 0.977 | 0.458 | 0.867 | 0.451 |
| + + + | 0.511 | 0.97 | 0.488 | 0.885 | 0.488 | |
| + + + | 0.539 | 0.962 | 0.475 | 0.888 | 0.488 | |
| + + + + | 0.539 | 0.961 | 0.47 | 0.886 | 0.488 | |
| 5.0 Å | + + | 0.395 | 0.98 | 0.488 | 0.858 | 0.530 |
| + + + | 0.48 | 0.968 | 0.511 | 0.874 | 0.563 | |
| + + + | 0.506 | 0.962 | 0.51 | 0.873 | 0.560 | |
| + + + + | 0.499 | 0.96 | 0.498 | 0.871 | 0.555 |
| Protein | Feature | Sensitivity | Specificity | MCC |
|---|---|---|---|---|
| MYC | + + | 0.30 | 0.941 | 0.282 |
| + + + | 0.70 | 0.853 | 0.448 | |
| + + + | 0.70 | 0.853 | 0.448 | |
| + + + + | 0.70 | 0.868 | 0.470 | |
| MAX | + + | 0.222 | 1.0 | 0.447 |
| + + + | 0.888 | 0.906 | 0.664 | |
| + + + | 0.888 | 0.922 | 0.697 | |
| + + + + | 0.889 | 0.922 | 0.697 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dang, T.K.L.; Meckbach, C.; Tacke, R.; Waack, S.; Gültas, M. A Novel Sequence-Based Feature for the Identification of DNA-Binding Sites in Proteins Using Jensen–Shannon Divergence. Entropy 2016, 18, 379. https://doi.org/10.3390/e18100379
Dang TKL, Meckbach C, Tacke R, Waack S, Gültas M. A Novel Sequence-Based Feature for the Identification of DNA-Binding Sites in Proteins Using Jensen–Shannon Divergence. Entropy. 2016; 18(10):379. https://doi.org/10.3390/e18100379
Chicago/Turabian StyleDang, Truong Khanh Linh, Cornelia Meckbach, Rebecca Tacke, Stephan Waack, and Mehmet Gültas. 2016. "A Novel Sequence-Based Feature for the Identification of DNA-Binding Sites in Proteins Using Jensen–Shannon Divergence" Entropy 18, no. 10: 379. https://doi.org/10.3390/e18100379
APA StyleDang, T. K. L., Meckbach, C., Tacke, R., Waack, S., & Gültas, M. (2016). A Novel Sequence-Based Feature for the Identification of DNA-Binding Sites in Proteins Using Jensen–Shannon Divergence. Entropy, 18(10), 379. https://doi.org/10.3390/e18100379