Unraveling of a Strongly Correlated Dynamical Network of Residues Controlling the Permeation of Potassium in KcsA Ion Channel


Abstract
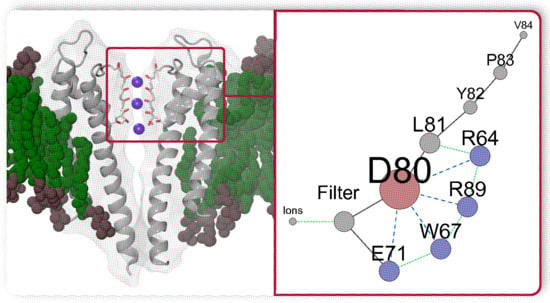
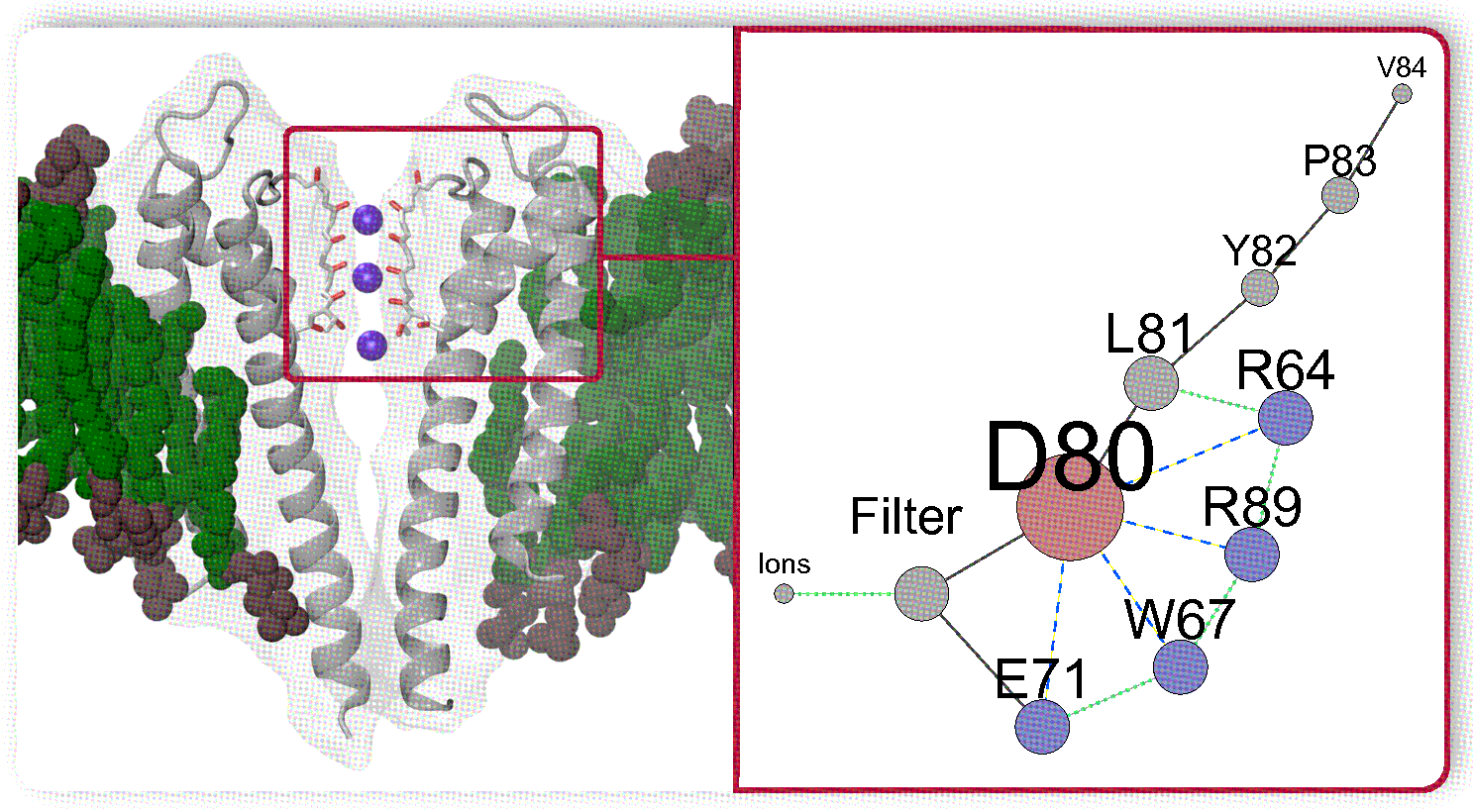
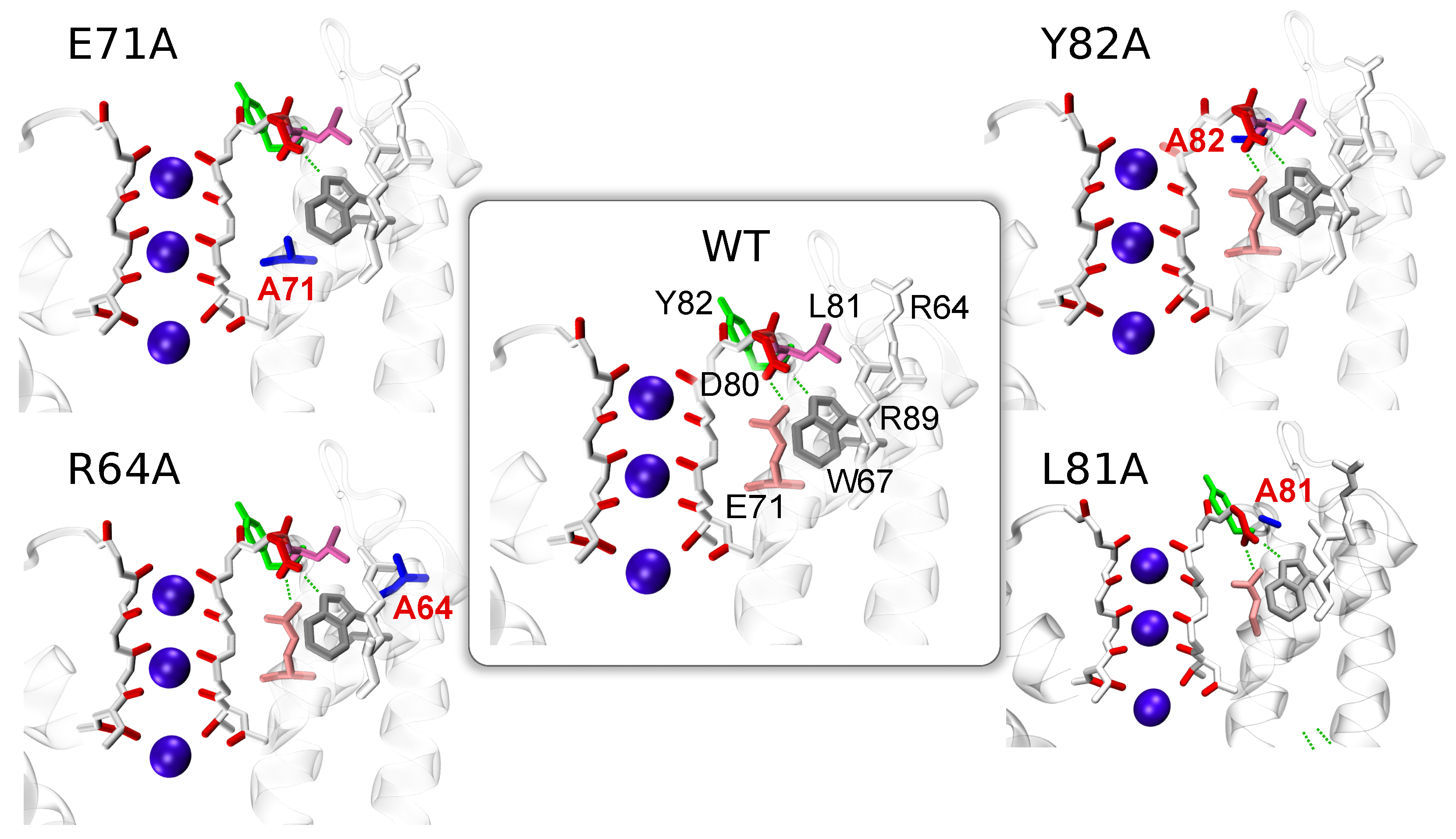
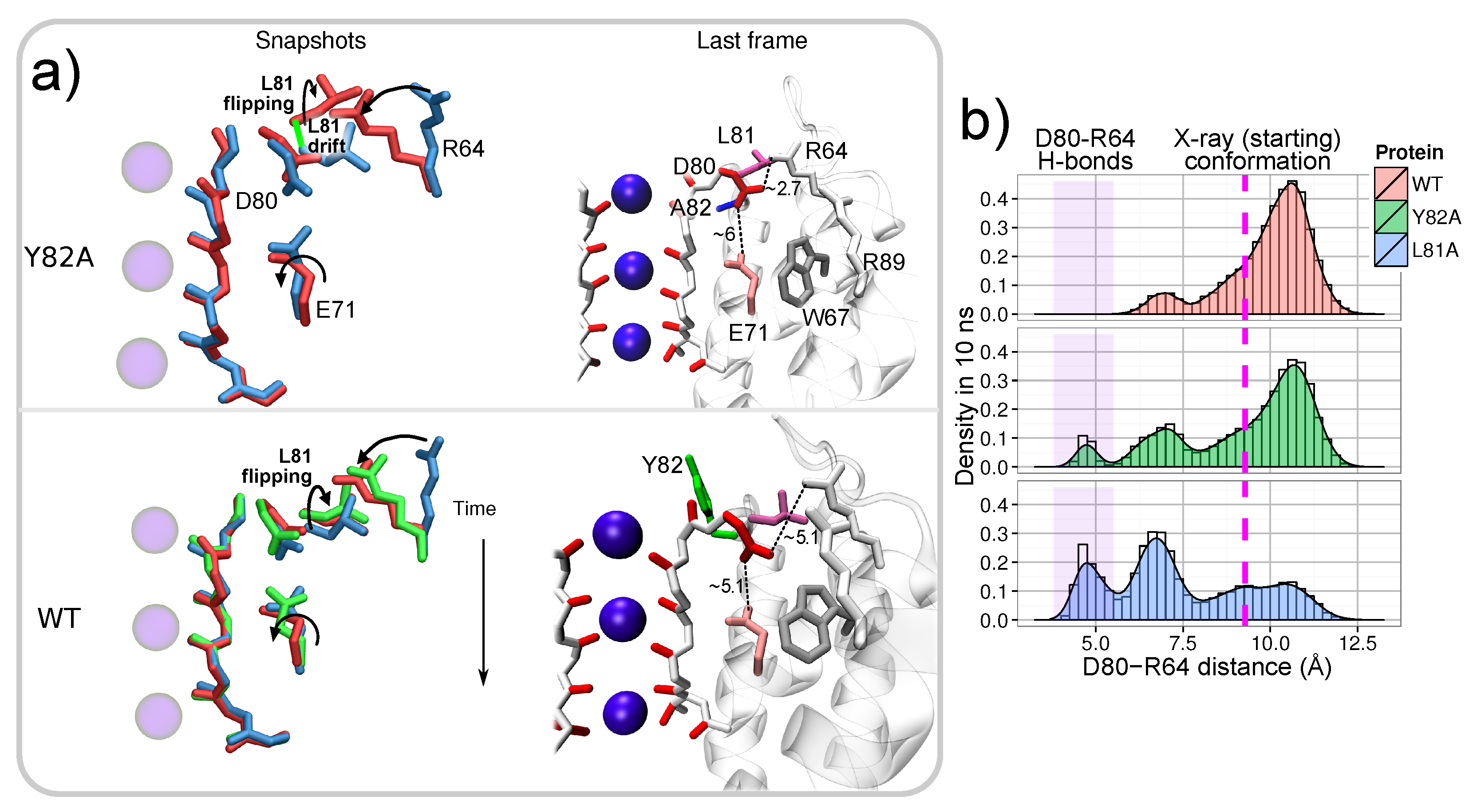
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Setup of the Simulations
2.2. Collective Variables and Order Parameters
2.3. Free Energy Calculations—Metadynamics
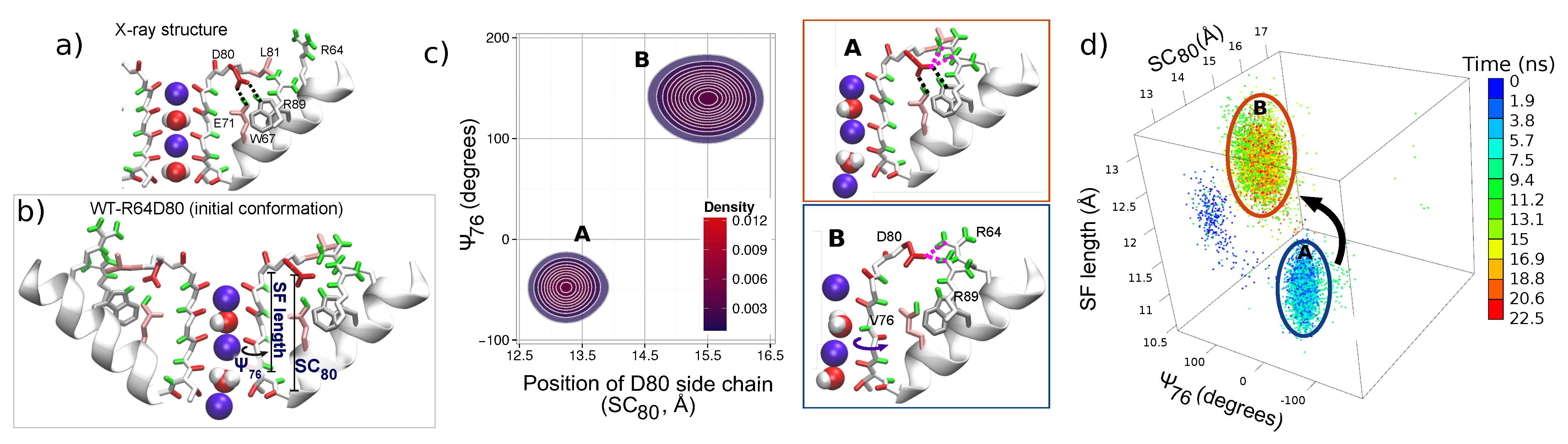
2.4. Initialization of WT-R64D80 Simulation
2.5. Statistical Analysis
3. Comparative Analysis of Dynamics of WT and Mutated Proteins
3.1. Considered Proteins
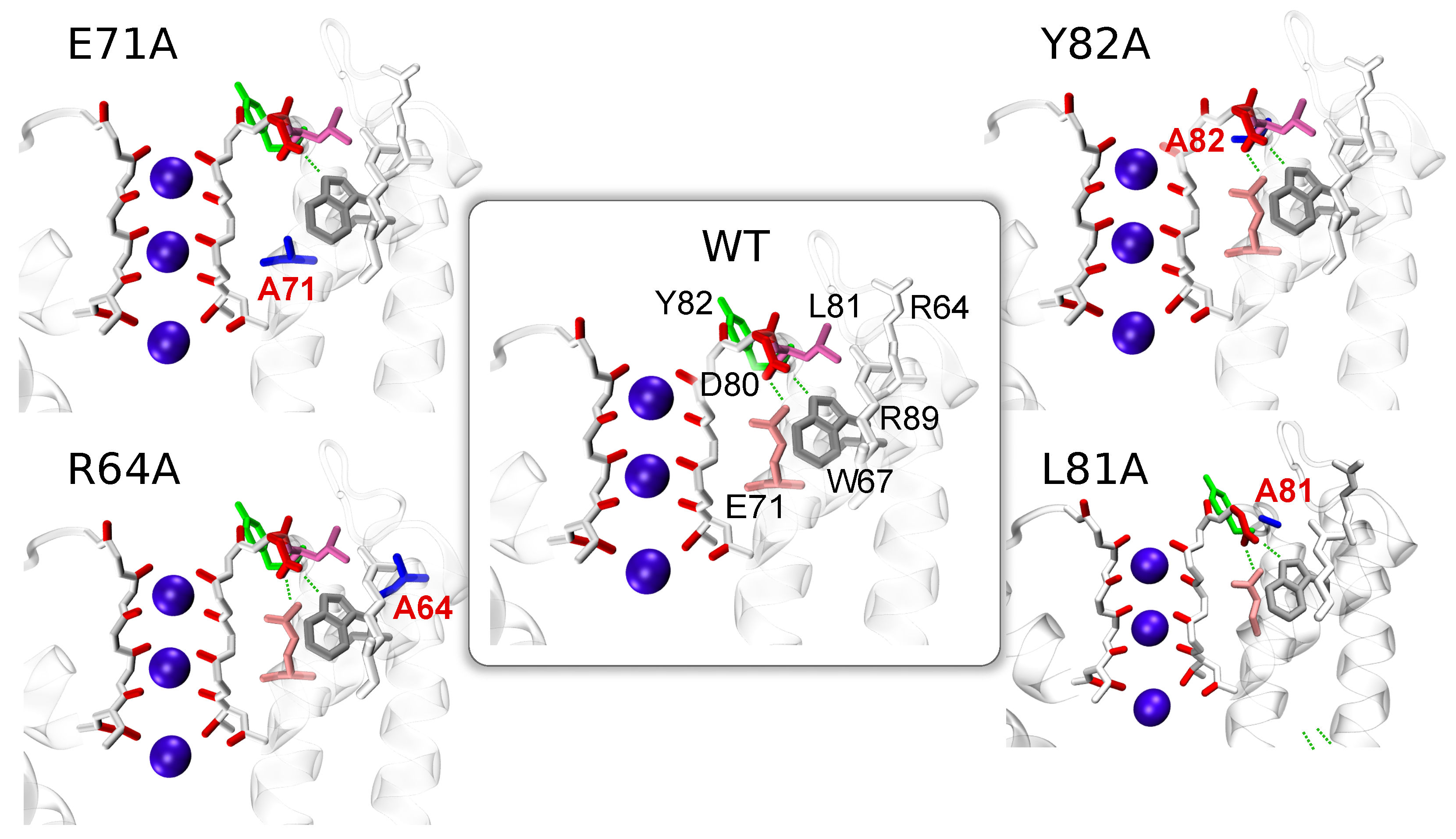
3.2. Dynamics of Mutant E71A
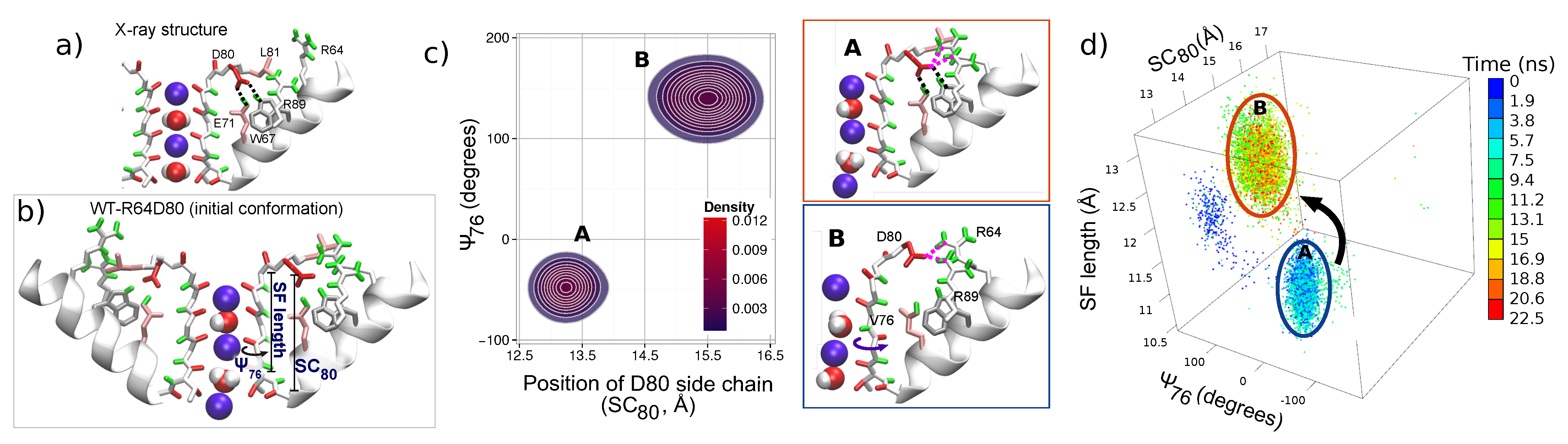
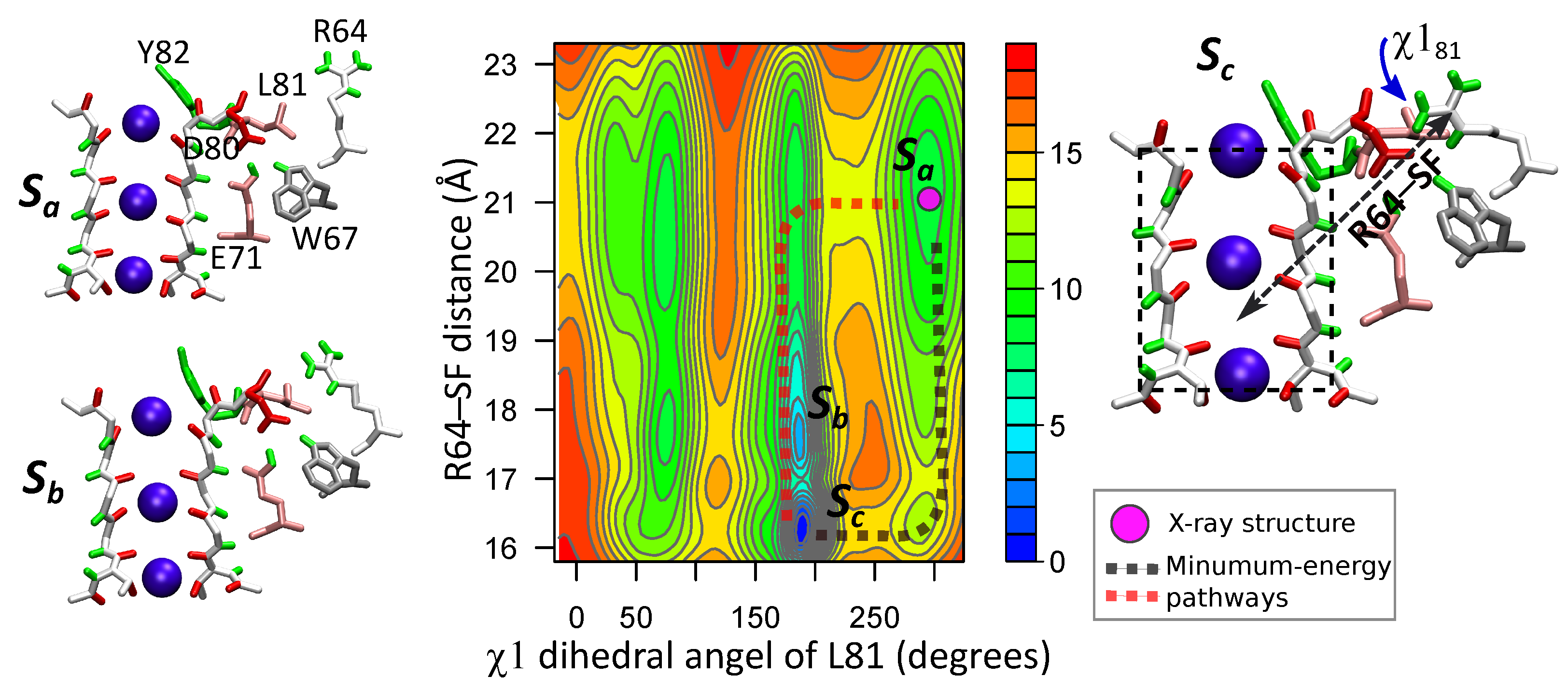
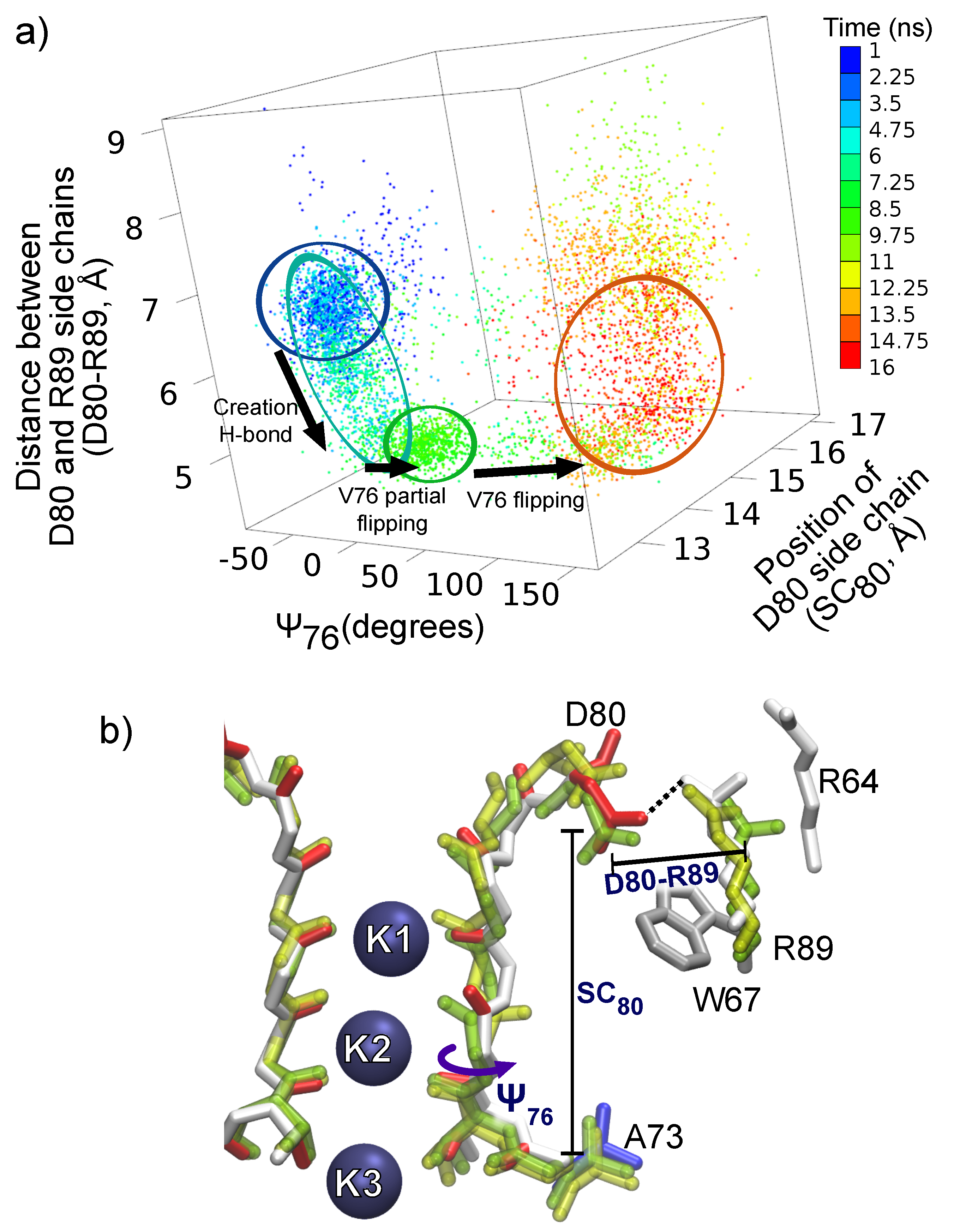
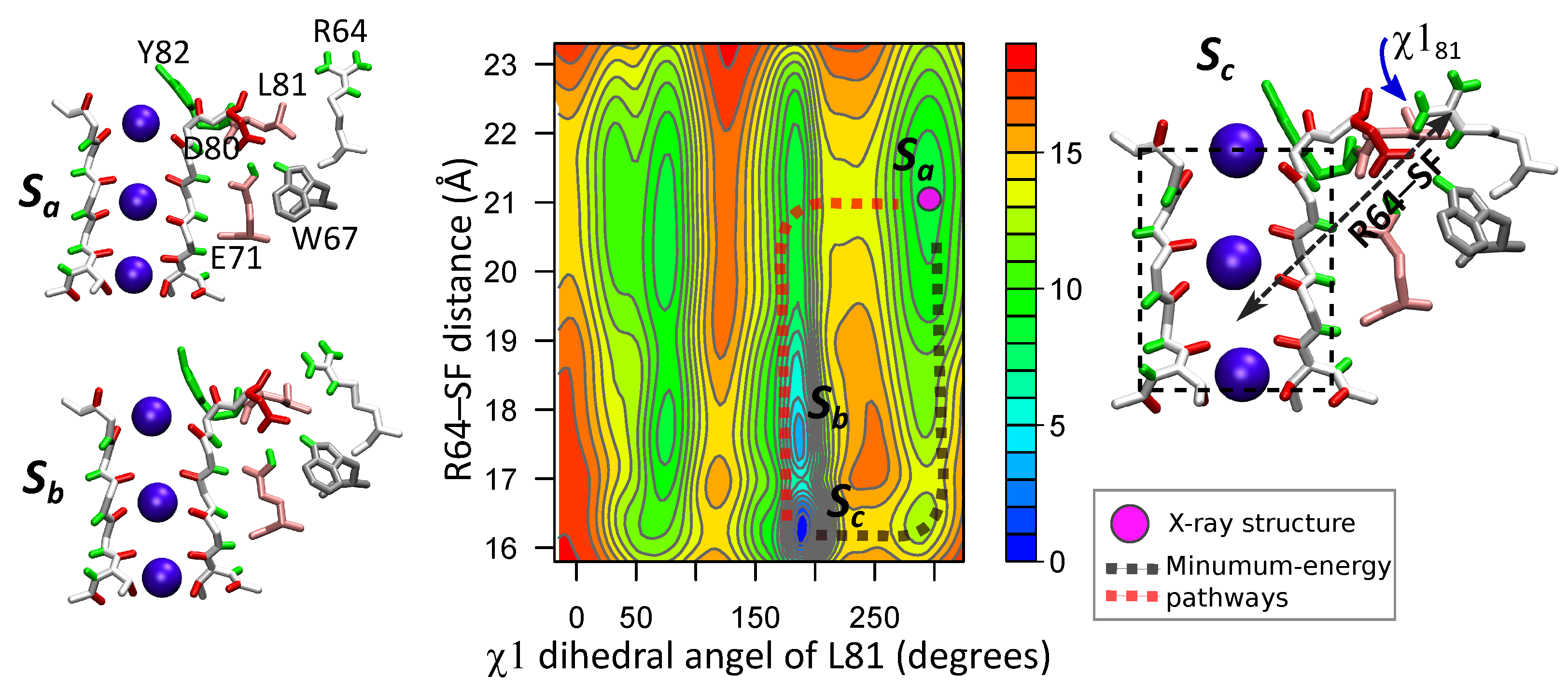
3.3. Correlated Dynamics of L81 and R64 Residues
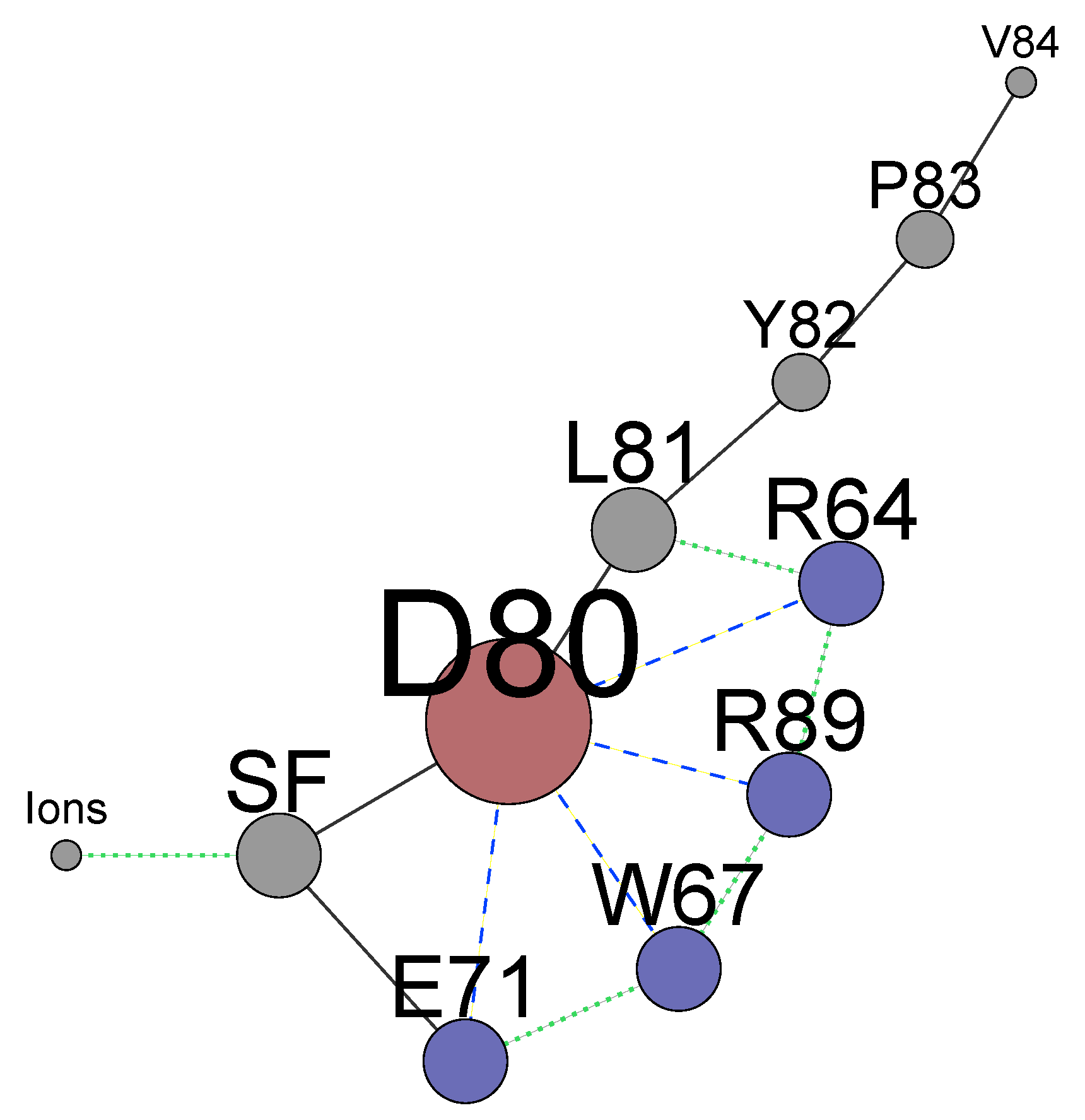
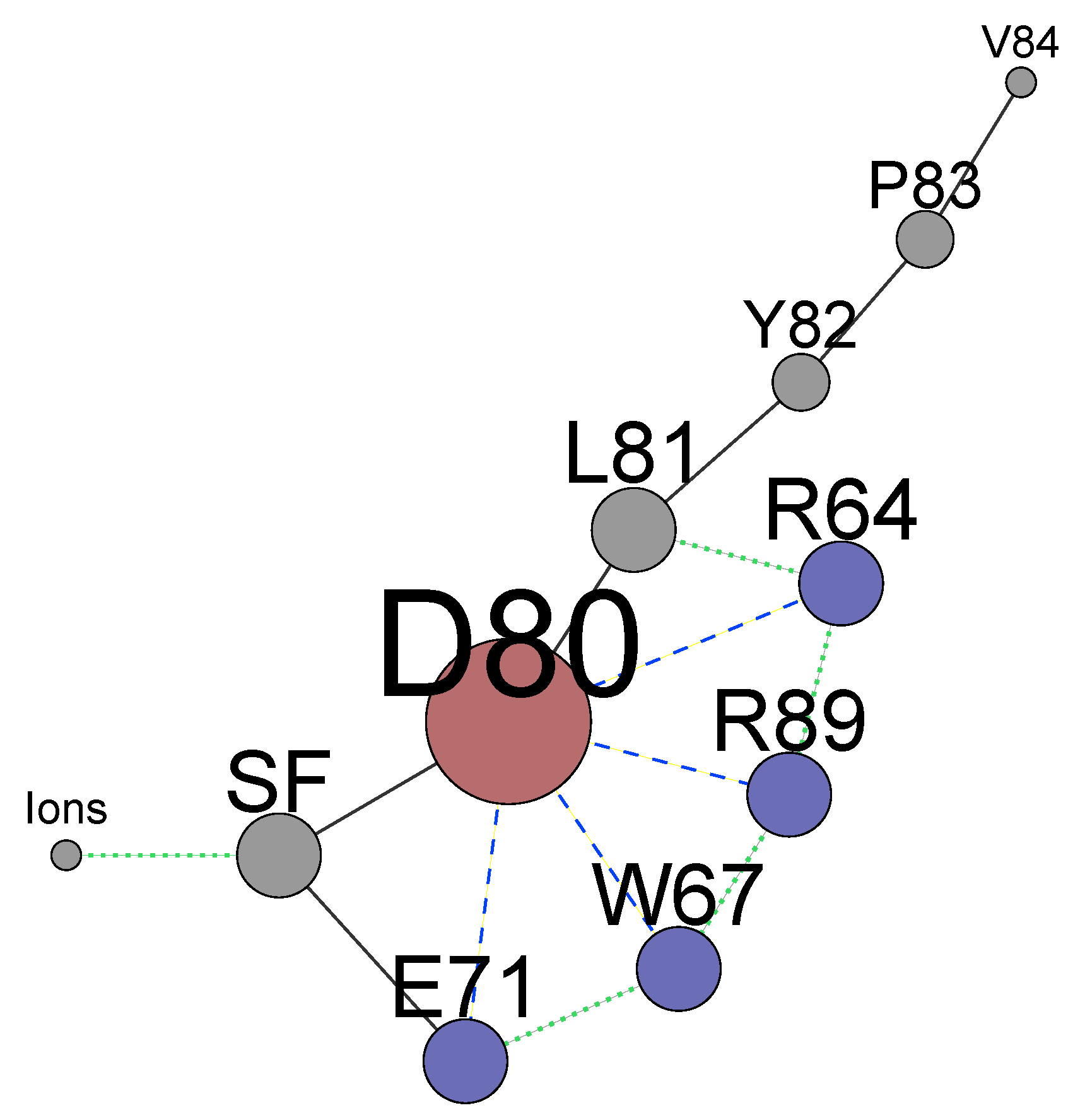
4. The Interactions of Residues and Ions in the WT Protein
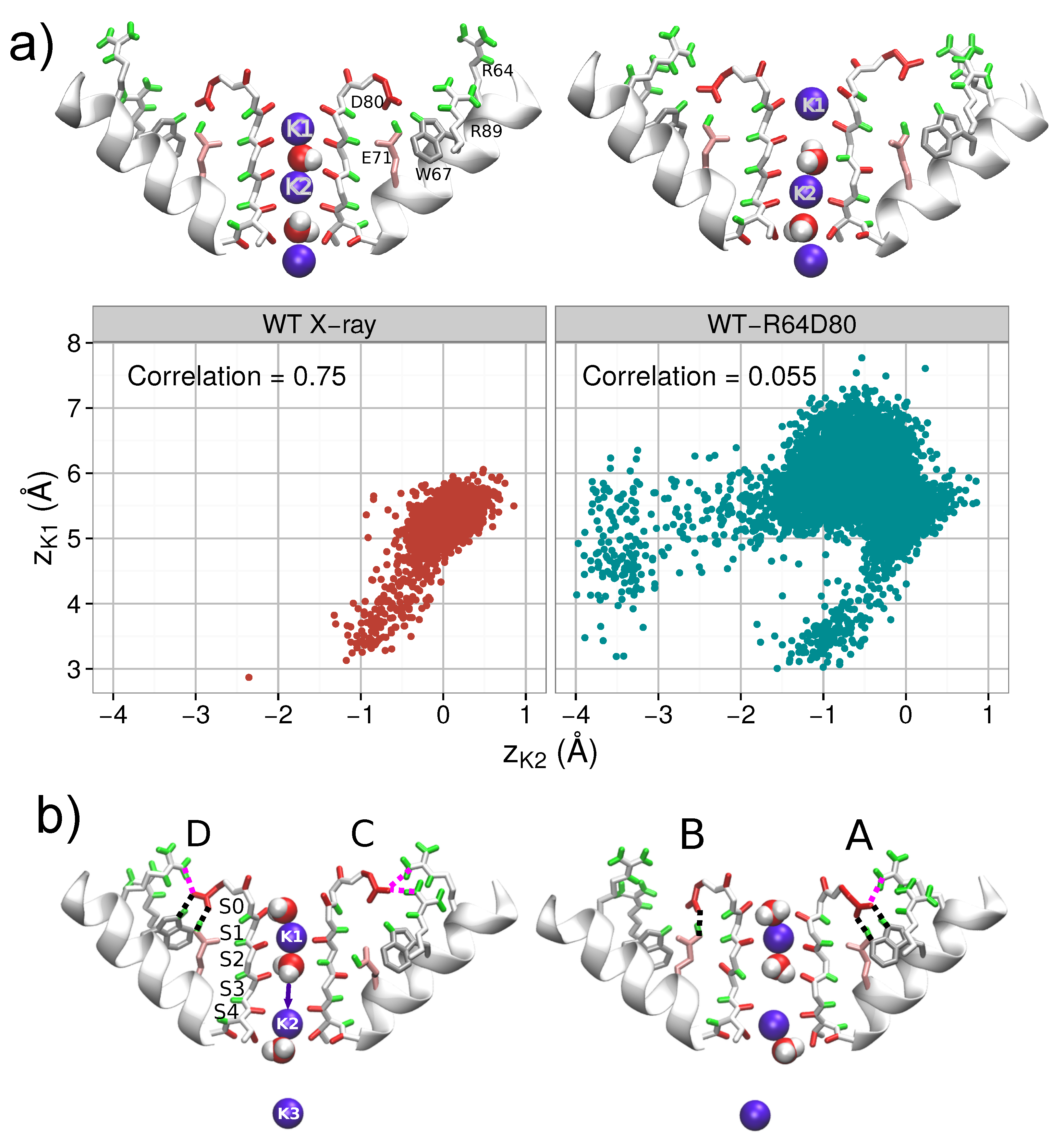
4.1. Influence of Arginines R64 and R89 on D80, the SF and Ions
4.2. Energetics of the Arginine Motions
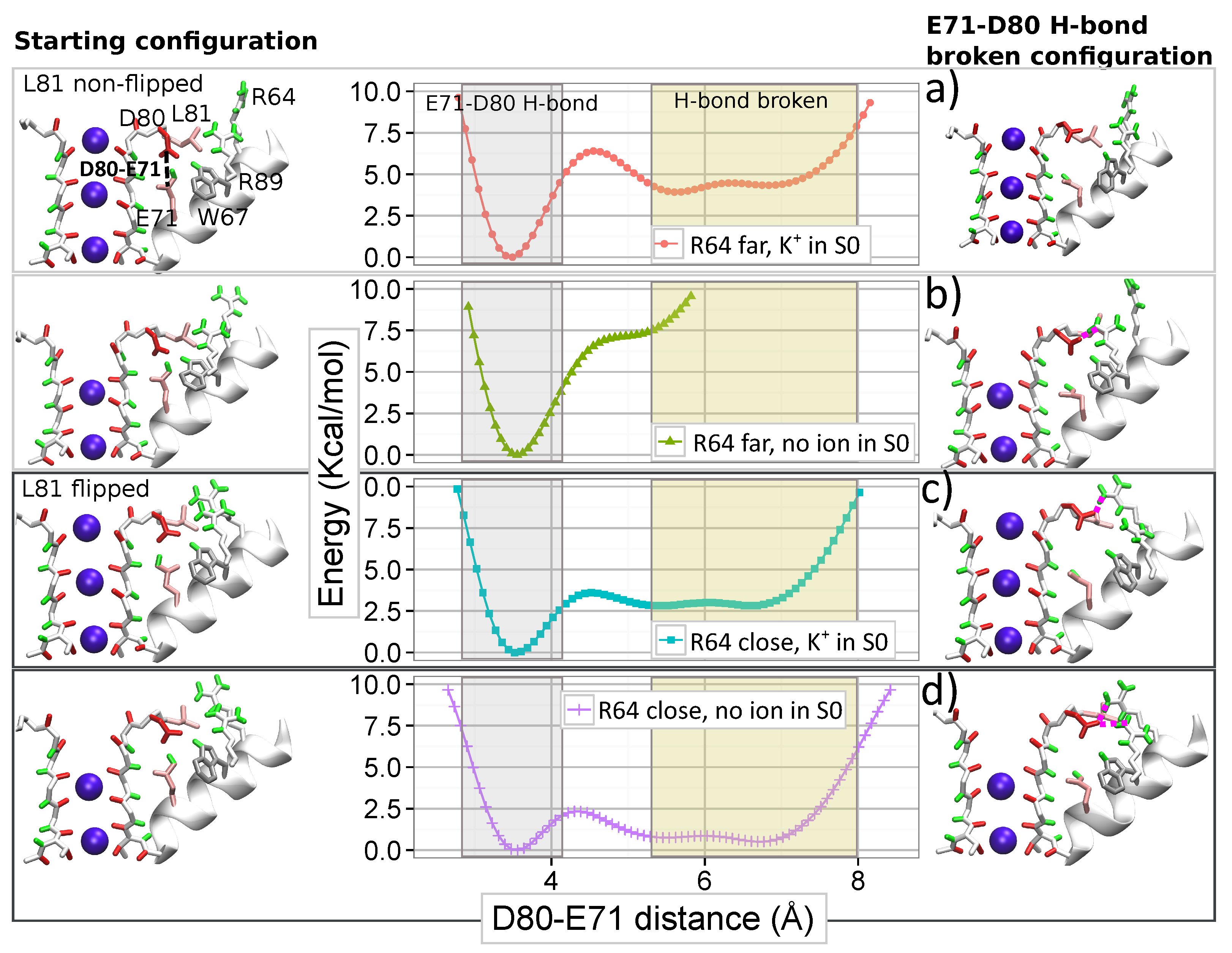
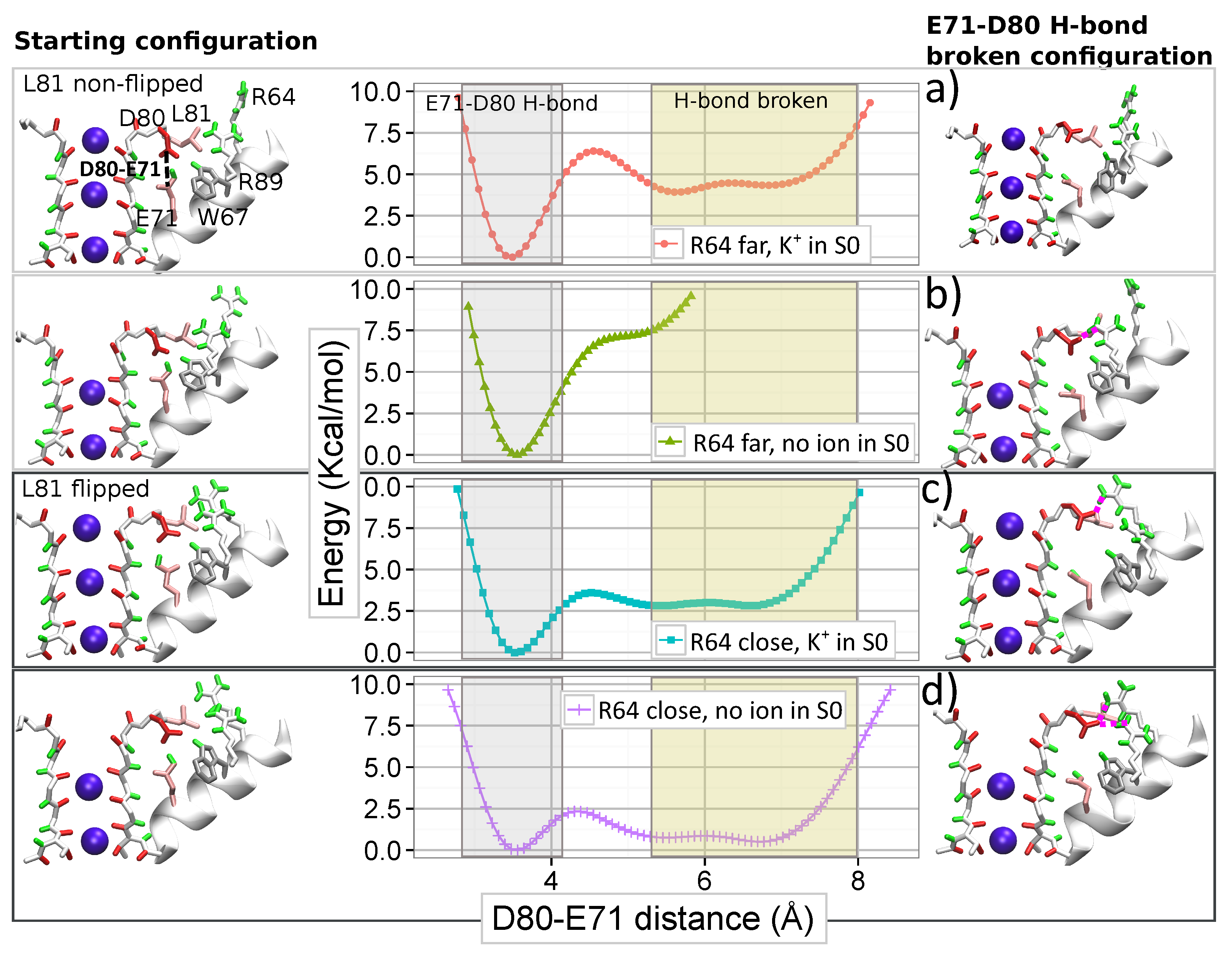
4.3. Opposite Influence of R64 and a K Ion Bound to S0 on the E71–D80 H-bond
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, M.; Morais-Cabral, J.H.; Mann, S.; MacKinnon, R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 2001, 411, 657–661. [Google Scholar] [CrossRef]
- Kratochvil, H.T.; Carr, J.K.; Matulef, K.; Annen, A.W.; Li, H.; Maj, M.; Ostmeyer, J.; Serrano, A.L.; Raghuraman, H.; Moran, S.D.; et al. Instantaneous ion configurations in the K+ ion channel selectivity filter revealed by 2D IR spectroscopy. Science 2016, 353, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopec, W.; Koepfer, D.A.; Vickery, O.N.; Bondarenko, A.S.; Jansen, T.L.C.; de Groot, B.L.; Zachariae, U. Direct knock-on of desolvated ions governs strict ion selectivity in K+ channels. Nat. Chem. 2018, 10, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMarco, K.R.; Bekker, S.; Vorobyov, I. Challenges and advances in atomistic simulations of potassium and sodium ion channel gating and permeation. J. Physiol. 2019, 597, 679–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumikama, T.; Oiki, S. Queueing arrival and release mechanism for K+ permeation through a potassium channel. J. Physiol. Sci. 2019, 69, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; McDermott, A.E. Inactivation in the potassium channel KcsA. J. Struct. Biol. X 2019, 3, 100009. [Google Scholar] [CrossRef]
- Renart, M.; Giudici, A.; Diaz-Garcia, C.; Molina, M.; Morales, A.; Gonzalez-Ros, J.; Poveda, J. Modulation of Function, Structure and Clustering of K+ Channels by Lipids: Lessons Learnt from KcsA. J. Mol. Sci. 2020, 21, 2554. [Google Scholar] [CrossRef] [Green Version]
- Strong, S.E.; Hestand, N.J.; Kananenka, A.A.; Zanni, M.T.; Skinner, J.L. IR Spectroscopy Can Reveal the Mechanism of K+ Transport in Ion Channels. Biophys. J. 2020, 118, 254–261. [Google Scholar] [CrossRef]
- Cordero-Morales, J.F.; Jogini, V.; Lewis, A.; Vasquez, V.; Cortes, D.M.; Roux, B.; Perozo, E. Molecular driving forces determining potassium channel slow inactivation. Nat. Struct. Mol. Biol. 2007, 14, 1062–1069. [Google Scholar] [CrossRef]
- Hoshi, T.; Armstrong, C.M. C-type inactivation of voltage-gated K+ channels: Pore constriction or dilation? J. Gen. Physiol. 2013, 141, 151–160. [Google Scholar] [CrossRef]
- Cuello, L.G.; Cortes, D.M.; Perozo, E. The gating cycle of a K+ channel at atomic resolution. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Chakrapani, S.; Cordero-Morales, J.F.; Perozo, E. A Quantitative Description of KcsA Gating I: Macroscopic Currents. J. Gen. Physiol. 2007, 130, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrapani, S.; Cordero-Morales, J.F.; Perozo, E. A Quantitative Description of KcsA Gating II: Single-Channel Currents. J. Gen. Physiol. 2007, 130, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Cordero-Morales, J.F.; Cuello, L.G.; Zhao, Y.; Jogini, V.; Cortes, D.M.; Roux, B.; Perozo, E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 2006, 13, 311–318. [Google Scholar] [CrossRef]
- Cordero-Morales, J.F.; Cuello, L.G.; Perozo, E. Voltage-dependent gating at the KcsA selectivity filter. Nat. Struct. Mol. Biol. 2006, 13, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Kurata, H.T.; Fedida, D. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 2006, 92, 185–208. [Google Scholar] [CrossRef]
- Raghuraman, H.; Islam, S.M.; Mukherjee, S.; Roux, B.; Perozo, E. Dynamics transitions at the outer vestibule of the KcsA potassium channel during gating. Proc. Natl. Acad. Sci. USA 2014, 111, 1831–1836. [Google Scholar] [CrossRef] [Green Version]
- Lockless, S.W.; Zhou, M.; MacKinnon, R. Structural and Thermodynamic Properties of Selective Ion Binding in a K+ Channel. PLoS Biol. 2007, 5, e121. [Google Scholar] [CrossRef]
- Renart, M.L.; Montoya, E.; Fernández, A.M.; Molina, M.L.; Poveda, J.A.; Encinar, J.A.; Ayala, J.L.; Ferrer-Montiel, A.V.; Gómez, J.; Morales, A.; et al. Contribution of Ion Binding Affinity to Ion Selectivity and Permeation in KcsA, a Model Potassium Channel. Biochemistry 2012, 51, 3891–3900. [Google Scholar] [CrossRef]
- Marius, P.; Zagnoni, M.; Sandison, M.E.; East, J.M.; Morgan, H.; Lee, A.G. Binding of Anionic Lipids to at Least Three Nonannular Sites on the Potassium Channel KcsA is Required for Channel Opening. Biophys. J. 2008, 94, 1689–1698. [Google Scholar] [CrossRef] [Green Version]
- Van der Cruijsen, E.A.W.; Nand, D.; Weingarth, M.; Prokofyev, A.; Hornig, S.; Cukkemane, A.A.; Bonvin, A.M.J.J.; Becker, S.; Hulse, R.E.; Perozo, E.; et al. Importance of lipid–pore loop interface for potassium channel structure and function. Proc. Natl. Acad. Sci. USA 2013, 110, 13008–13013. [Google Scholar] [PubMed] [Green Version]
- Radda, R.; Kim, D.; Nimigean, C.; Andersen, O. Regulation of Ion Channel Function by the Host Lipid Bilayer Examined by a Stopped-Flow Spectrofluorometric Assay. Biophys. J. 2014, 106, 1070–1078. [Google Scholar]
- Poveda, J.; Giudici, A.M.; Renart, M.L.; Millet, O.; Morales, A.; Gonzalez-Ros, J.M.; Oakes, V.; Furini, S.; Domene, C. Modulation of the potassium channel KcsA by anionic phospholipids: Role of arginines at the non-annular lipid binding sites. Biochim. Biophys. Acta (BBA) Biomembr. 2019, 1861, 183029. [Google Scholar]
- Hille, B. Ion Channels of Excitable Membranes; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perozo, E.; Marien, C.D.; Cuello, L.G. Structural Rearrangements Underlying K+-Channel Activation Gating. Science 1999, 285, 73–78. [Google Scholar] [PubMed] [Green Version]
- Takeuchi, K.; Takahashi, H.; Kawano, S.; Shimada, I. Identification and Characterization of the Slowly Exchanging pH-dependent Conformational Rearrangement in KcsA. J. Biol. Chem. 2007, 282, 15179–15186. [Google Scholar]
- Zhou, Y.; Morais-Cabral, J.H.; Kaufman, A.; MacKinnon, R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature 2001, 414, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Morales, J.F.; Jogini, V.; Chakrapani, S.; Perozo, E. A Multipoint Hydrogen-Bond Network Underlying KcsA C-Type Inactivation. Biophys. J. 2011, 100, 2387–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrapani, S.; Cordero-Morales, J.F.; Jogini, V.; Pan, A.C.; Cortes, D.M.; Roux, B.; Perozo, E. On the structural basis of modal gating behavior in K+ channels. Nat. Struct. Mol. Biol. 2010, 18, 67–74. [Google Scholar]
- Chapman, M.L.; Krovetz, H.S.; VanDongen, A.M.J. GYGD pore motifs in neighbouring potassium channel subunits interact to determine ion selectivity. J. Physiol. 2001, 530, 21–33. [Google Scholar] [CrossRef]
- Cheng, W.W.L.; McCoy, J.G.; Thompson, A.N.; Nichols, C.G.; Nimigean, C.M. Mechanism for selectivity-inactivation coupling in KcsA potassium channels. Proc. Natl. Acad. Sci. USA 2011, 108, 5272–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marius, P.; de Planque, M.R.R.; Williamson, P.T.F. Probing the interaction of lipids with the non-annular binding sites of the potassium channel KcsA by magic-angle spinning NMR. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florian, T.H.; David, J.P.; Wojciech, W.N.; Crina, M.N.; Berneche, S. Mechanism of activation at the selectivity filter of the KcsA K+ channel. eLife 2017, 6, e25844. [Google Scholar]
- Labro, A.J.; Cortes, D.M.; Tilegenova, C.; Cuello, L.G. Inverted allosteric coupling between activation and inactivation gates in K+ channels. Proc. Natl. Acad. Sci. USA 2018, 115, 5426–5431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Xu, Y.; Zhang, D.; McDermott, A.E. Probing allosteric coupling in a constitutively open mutant of the ion channel KcsA using solid-state NMR. Proc. Natl. Acad. Sci. USA 2020, 117, 7171–7175. [Google Scholar] [PubMed]
- Devaraneni, P.K.; Komarov, A.G.; Costantino, C.A.; Devereaux, J.J.; Matulef, K.; Valiyaveetil, F.I. Semisynthetic K+ channels show that the constricted conformation of the selectivity filter is not the C-type inactivated state. Proc. Natl. Acad. Sci. USA 2013, 110, 15698–15703. [Google Scholar] [CrossRef] [Green Version]
- Matulef, K.; Komarov, A.G.; Costantino, C.A.; Valiyaveetil, F.I. Using protein backbone mutagenesis to dissect the link between ion occupancy and C-type inactivation in K+ channels. Proc. Natl. Acad. Sci. USA 2013, 110, 17886–17891. [Google Scholar] [CrossRef] [Green Version]
- Matulef, K.; Annen, A.; Nix, J.; Valiyaveetil, F. Individual Ion Binding Sites in the K+ Channel Play Distinct Roles in C-type Inactivation and in Recovery from Inactivation. Structure 2016, 24, 750–761. [Google Scholar] [CrossRef]
- Baker, K.A.; Tzitzilonis, C.; Kwiatkowski, W.; Choe, S.; Riek, R. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat. Struct. Mol. Biol. 2007, 14, 1089–1095. [Google Scholar] [CrossRef]
- Jekhmane, S.; Medeiros-Silva, J.; Li, J.; Kummerer, F.; Muller-Hermes, C.; Baldus, M.; Roux, B.; Weingarth, M. Shifts in the selectivity filter dynamics cause modal gating in K+ channels. Nat. Commun. 2019, 10, 123. [Google Scholar]
- Li, J.; Ostmeyer, J.; Boulanger, E.; Rui, H.; Perozo, E.; Roux, B. Chemical substitutions in the selectivity filter of potassium channels do not rule out constricted-like conformations for C-type inactivation. Proc. Natl. Acad. Sci. USA 2017, 114, 11145–11150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swenson, R.P.; Armstrong, C.M. K+ channels close more slowly in the presence of external K+ and Rb+. Nature 1981, 291, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Piasta, K.N.; Theobald, D.L.; Miller, C. Potassium-selective block of barium permeation through single KcsA channels. J. Gen. Phys. 2011, 138, 421–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Focke, P.J.; Matulef, K.; Bian, X.; Moënne-Loccoz, P.; Valiyaveetil, F.I.; Lockless, S.W. Ion-binding properties of a K+ channel selectivity filter in different conformations. Proc. Natl. Acad. Sci. USA 2015, 112, 15096–15100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renart, M.L.; Giudici, A.M.; Poveda, J.A.; Fedorov, A.; Berberan-Santos, M.N.; Prieto, M.; Diaz-Garcia, C.; Gonzalez-Ros, J.M.; Coutinho, A. Conformational plasticity in the KcsA potassium channel pore helix revealed by homo-FRET studies. Sci. Rep. 2019, 9, 6215. [Google Scholar] [CrossRef] [PubMed]
- Sansom, M.S.; Shrivastava, I.H.; Bright, J.N.; Tate, J.; Capener, C.E.; Biggin, P.C. Potassium channels: Structures, models, simulations. Biochim. Biophys. Acta (BBA) Biomembr. 2002, 1565, 294–307. [Google Scholar] [CrossRef] [Green Version]
- Maffeo, C.; Bhattacharya, S.; Yoo, J.; Wells, D.; Aksimentiev, A. Modeling and Simulation of Ion Channels. Chem. Rev. 2012, 112, 6250–6284. [Google Scholar] [CrossRef] [Green Version]
- Harpole, T.J.; Delemotte, L. Conformational landscapes of membrane proteins delineated by enhanced sampling molecular dynamics simulations. Biochim. Biophys. Acta (BBA) Biomembr. 2018, 1860, 909–926. [Google Scholar] [CrossRef]
- Noskov, S.Y.; Bernèche, S.; Roux, B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature 2004, 431, 830–834. [Google Scholar] [CrossRef]
- Tilegenova, C.; Cortes, D.M.; Jahovic, N.; Hardy, E.; Hariharan, P.; Guan, L.; Cuello, L.G. Structure, function, and ion-binding properties of a K+ channel stabilized in the 2,4-ion–bound configuration. Proc. Natl. Acad. Sci. USA 2019, 116, 16829–16834. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ostmeyer, J.; Cuello, L.G.; Perozo, E.; Roux, B. Rapid constriction of the selectivity filter underlies C-type inactivation in the KcsA potassium channel. J. Gen. Physiol. 2018, 150, 1408–1420. [Google Scholar] [CrossRef] [Green Version]
- Delemotte, L. Opening leads to closing: Allosteric crosstalk between the activation and inactivation gates in KcsA. J. Gen. Physiol. 2018, 150, 1356–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furini, S.; Domene, C. Critical Assessment of Common Force Fields for Molecular Dynamics Simulations of Potassium Channels. J. Chem. Theory Comput. 2020, 16, 7148–7159. [Google Scholar] [CrossRef]
- Cuello, L.G.; Jogini, V.; Cortes, D.M.; Pan, A.C.; Gagnon, D.G.; Dalmas, O.; Cordero-Morales, J.F.; Chakrapani, S.; Roux, B.; Perozo, E. Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature 2010, 466, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Wojtas-Niziurski, W.; Meng, Y.; Roux, B.; Bernache, S. Self-Learning Adaptive Umbrella Sampling Method for the Determination of Free Energy Landscapes in Multiple Dimensions. J. Chem. Theory Comput. 2013, 9, 1885–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeMasurier, M.; Heginbotham, L.; Miller, C. Kcsa: It’s a Potassium Channel. J. Gen. Physiol. 2001, 118, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.W.; Abad, E.; Beckstein, O.; Sansom, M.S.P. Energetics of Multi-Ion Conduction Pathways in Potassium Ion Channels. J. Chem. Theory Comput. 2013, 9, 5176–5189. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.O.; Jogini, V.; Eastwood, M.P.; Shaw, D.E. Atomic-level simulation of current-voltage relationships in single-file ion channels. J. Gen. Physiol. 2013, 141, 619–632. [Google Scholar] [PubMed]
- Köpfer, D.A.; Song, C.; Gruene, T.; Sheldrick, G.M.; Zachariae, U.; de Groot, B.L. Ion permeation in K+ channels occurs by direct Coulomb knock-on. Science 2014, 346, 352–355. [Google Scholar] [CrossRef] [Green Version]
- Sumikama, T.; Oiki, S. Digitalized K+ Occupancy in the Nanocavity Holds and Releases Queues of K+ in a Channel. J. Am. Chem. Soc. 2016, 138, 10284–10292. [Google Scholar]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosseddu, S.M. Structure and Dynamics of Protein in the Permeation and Gating of Potassium Ion Channels: Identifying Molecular Determinants and Developing Coarse-Grained Approaches. Ph.D. Thesis, University of Warwick, Coventry, UK, 2013. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-range Interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A.D.; Wiorkiewicz-Kuczera, J.; Karplus, M. An all-atom empirical energy function for the simulation of nucleic acids. J. Am. Chem. Soc. 1995, 117, 11946–11975. [Google Scholar] [CrossRef]
- Buck, M.; Bouguet-Bonnet, S.; Pastor, R.W.; MacKerell, A.D. Importance of the CMAP Correction to the CHARMM22 Protein Force Field: Dynamics of Hen Lysozyme. Biophys. J. 2006, 90, L36–L38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Bernèche, S.; Roux, B. The Ionization State and the Conformation of Glu-71 in the KcsA K+ Channel. Biophys. J. 2002, 82, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Bucher, D.; Guidoni, L.; Rothlisberger, U. The Protonation State of the Glu-71/Asp-80 Residues in the KcsA Potassium Channel: A First-Principles QM/MM Molecular Dynamics Study. Biophys. J. 2007, 93, 2315–2324. [Google Scholar] [CrossRef] [Green Version]
- Grubmüller, H.; Groll, V. Solvate. Available online: https://www.mpibpc.mpg.de/grubmueller/solvate (accessed on 4 January 2021).
- Zhang, L.; Hermans, J. Hydrophilicity of cavities in proteins. Proteins Struct. Funct. Bioinform. 1996, 24, 433–438. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Fiorin, G.; Klein, M.L.; Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 2013, 111, 3345–3362. [Google Scholar] [CrossRef]
- Humphrey, W. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Adler, D.; Murdoch, D. rgl: 3D Visualization Device System (OpenGL), R Package Version 0.92.892; Available online: http://r-forge.r-project.org/projects/rgl (accessed on 5 January 2021).
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 2nd ed.; Sage: Thousand Oaks, CA, USA, 2011. [Google Scholar]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Furrer, R.; Nychka, D.; Sain, S. Fields: Tools for Spatial Data, R Package Version 6.7.6; Available online: https://cran.r-project.org/src/contrib/Archive/fields (accessed on 5 January 2021).
- Berneche, S.; Roux, B. Energetics of ion conduction through the K+ channel. Nature 2001, 414, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Bernèche, S.; Roux, B. Molecular Dynamics of the KcsA K+ Channel in a Bilayer Membrane. Biophys. J. 2000, 78, 2900–2917. [Google Scholar] [CrossRef] [Green Version]
- Bernèche, S.; Roux, B. A Gate in the Selectivity Filter of Potassium Channels. Structure 2005, 13, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Domene, C.; Klein, M.L.; Branduardi, D.; Gervasio, F.L.; Parrinello, M. Conformational Changes and Gating at the Selectivity Filter of Potassium Channels. J. Am. Chem. Soc. 2008, 130, 9474–9480. [Google Scholar] [CrossRef]
- Piccinini, E.; Ceccarelli, M.; Affinito, F.; Brunetti, R.; Jacoboni, C. Biased Molecular Simulations for Free-Energy Mapping: A Comparison on the KcsA Channel as a Test Case. J. Chem. Theory Comput. 2008, 4, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Miloshevsky, G.V.; Jordan, P.C. Conformational Changes in the Selectivity Filter of the Open-State KcsA Channel: An Energy Minimization Study. Biophys. J. 2008, 95, 3239–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, I.H.; Sansom, M.S.P. Simulations of Ion Permeation Through a Potassium Channel: Molecular Dynamics of KcsA in a Phospholipid Bilayer. Biophys. J. 2000, 78, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Compoint, M.; Carloni, P.; Ramseyer, C.; Girardet, C. Molecular dynamics study of the KcsA channel at 2.0-A resolution: Stability and concerted motions within the pore. Biochim. Biophys. Acta Biomem. 2004, 1661, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Allen, T.W. On the selective ion binding hypothesis for potassium channels. Proc. Natl. Acad. Sci. USA 2011, 108, 17963–17968. [Google Scholar] [CrossRef] [Green Version]
- Furini, S.; Domene, C. Atypical mechanism of conduction in potassium channels. Proc. Natl. Acad. Sci. USA 2009, 106, 16074–16077. [Google Scholar] [CrossRef] [Green Version]
- Deol, S.S.; Domene, C.; Bond, P.J.; Sansom, M.S. Anionic Phospholipid Interactions with the Potassium Channel KcsA: Simulation Studies. Biophys. J. 2006, 90, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Ion channel voltage sensors: Structure, function, and pathophysiology. Neuron 2010, 67, 915–928. [Google Scholar] [CrossRef] [Green Version]
- Gibby, W.A.T.; Barabash, M.L.; Guardiani, C.; Luchinsky, D.G.; McClintock, P.V.E. Physics of selective conduction and point mutation in biological ion channels. arXiv 2020, arXiv:physics.bio-ph/2010.08450. [Google Scholar]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosseddu, S.M.; Choe, E.J.; Khovanov, I.A. Unraveling of a Strongly Correlated Dynamical Network of Residues Controlling the Permeation of Potassium in KcsA Ion Channel. Entropy 2021, 23, 72. https://doi.org/10.3390/e23010072
Cosseddu SM, Choe EJ, Khovanov IA. Unraveling of a Strongly Correlated Dynamical Network of Residues Controlling the Permeation of Potassium in KcsA Ion Channel. Entropy. 2021; 23(1):72. https://doi.org/10.3390/e23010072
Chicago/Turabian StyleCosseddu, Salvatore M., Eunju Julia Choe, and Igor A. Khovanov. 2021. "Unraveling of a Strongly Correlated Dynamical Network of Residues Controlling the Permeation of Potassium in KcsA Ion Channel" Entropy 23, no. 1: 72. https://doi.org/10.3390/e23010072
APA StyleCosseddu, S. M., Choe, E. J., & Khovanov, I. A. (2021). Unraveling of a Strongly Correlated Dynamical Network of Residues Controlling the Permeation of Potassium in KcsA Ion Channel. Entropy, 23(1), 72. https://doi.org/10.3390/e23010072