
Prediction of sgRNA Off-Target Activity in CRISPR/Cas9 Gene Editing Using Graph Convolution Network



Abstract
:1. Introduction
- sgRNA (sequence of 20 nucleotides in length) needs to be complementary with its targeting genome sequence.
- Cas9 protein cleaves the target DNA at the site, three bases upstream of PAM, under the guidance of sgRNA sequence.
- to develop a graph-based approach for off-target prediction in the CRISPR/Cas9 system that is easy to understand and implement by researchers,
- to use a link prediction method and to predict the presence of links between sgRNA and off-target producing target sequences,
- to use a graph convolutional network (GCN), a powerful neural network model used for performing representation learning of network graphs created from off-target data set,
- to provide features for sequences (nodes) in the graph by extracting features from within the sequences,
- to handle the imbalance in the off-target dataset using cluster data sampling, by random sampling of sequences in every cluster in the network graph, and,
- to make use of StellarGraph [50] library that enables researchers to easily identify patterns and implement graph machine learning.
2. Background
2.1. Graph Convolutional Network
2.2. Link Prediction Using Stellargraph
3. Related Works
3.1. DeepCrispr
3.1.1. Architecture
3.1.2. Feature Extraction
3.1.3. Performance
3.1.4. Review
3.2. CNN_Std
3.2.1. Architecture
3.2.2. Feature Extraction
3.2.3. Performance
3.2.4. Review
3.3. AttnToMismatch_CNN
3.3.1. Architecture
3.3.2. Feature Extraction
3.3.3. Performance
3.3.4. Review
3.4. CnnCrispr
3.4.1. Architecture
3.4.2. Feature Extraction
3.4.3. Performance
3.4.4. Review
4. Materials and Methodology
4.1. Dataset
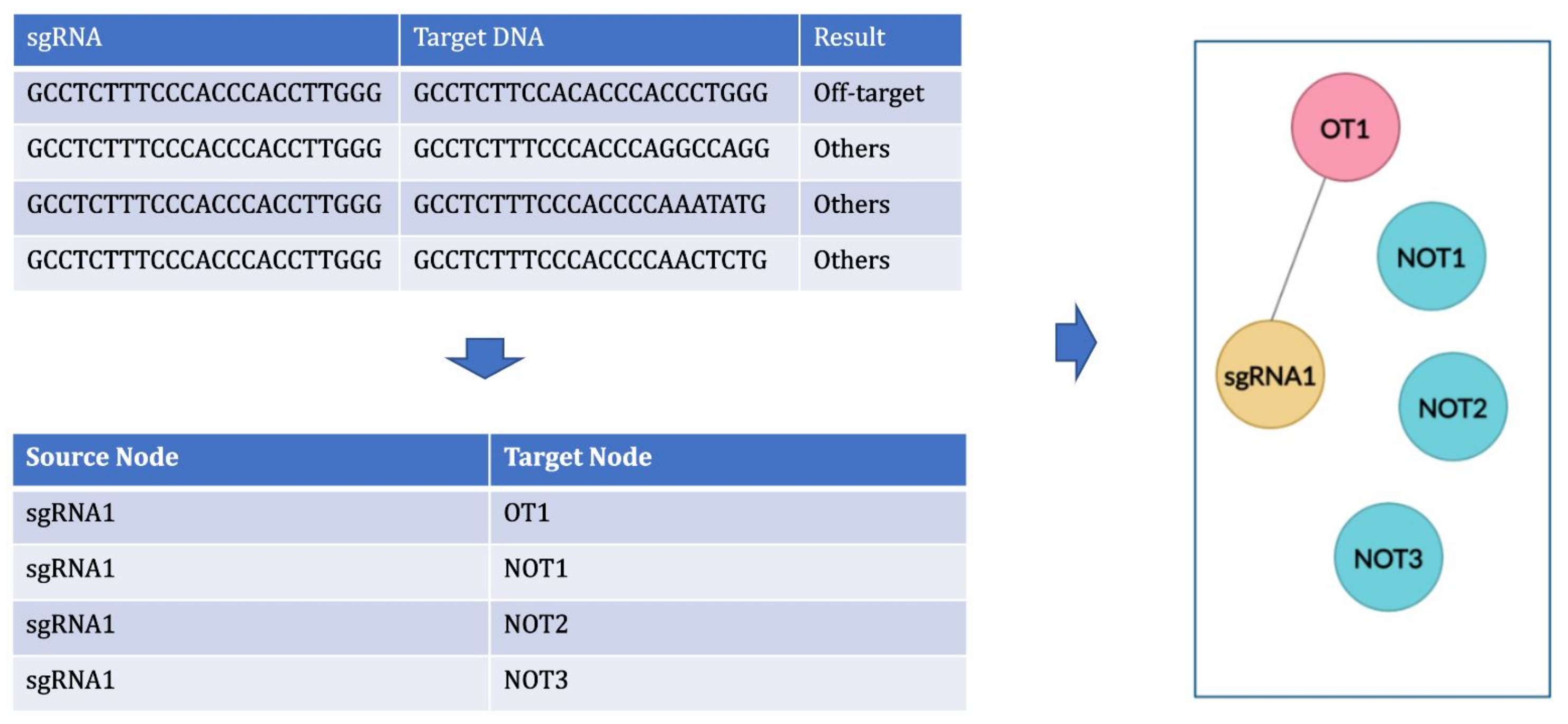
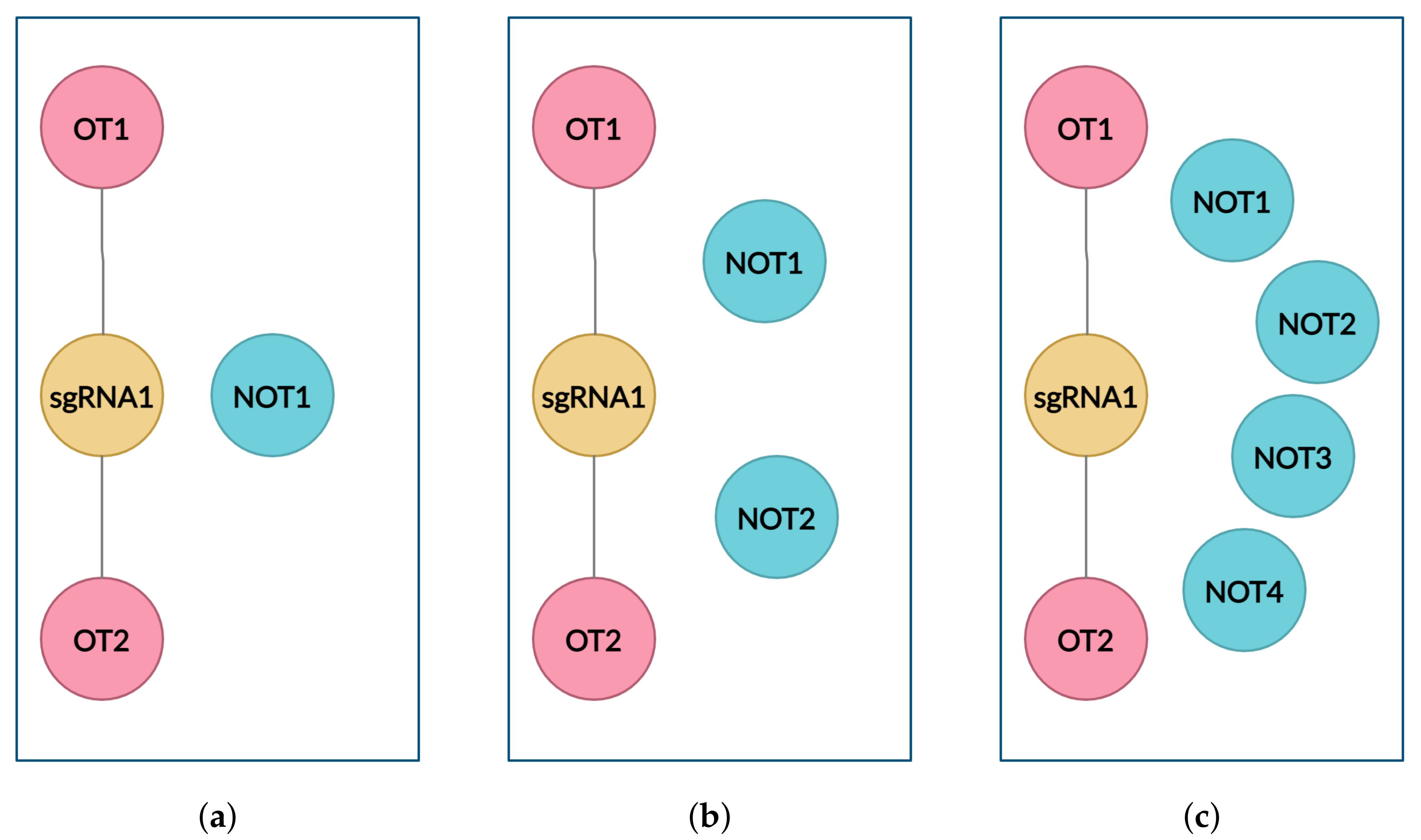
4.2. Graph
- to create the graph with only positive links (links between sgRNA and off-target inducing target DNA (OT) sequences);
- to create balanced clusters containing an equal number of OT and NOT sequences for every sgRNA cluster using cluster data sampling.
4.3. Cluster Data Sampling
4.4. Feature Extraction
4.4.1. Case Study 1: Nucleotide Occurrence
4.4.2. Case Study 2: Nucleotide Position
4.5. Graph Analysis
5. Results and Discussion
- A graph-based approach to predict off-target mutations in CRISPR/Cas9 gene editing, which is easy to implement and replicate by researchers, is possible. Link prediction is done to predict the presence of links between sgRNA and off-target inducing target DNA (OT) sequences.
- GCN, a powerful graph neural network model used for performing representation learning of graphs, can predict off-target efficacy in CRISPR/Cas9 gene editing by performing link prediction on the off-target dataset.
- Sequence-based features, such as position and occurrences of nucleotides in a sequence can increase the performance of GCN model in analysing graphs with high accuracy. GCN is used to validate the off-target efficacy of sgRNA in both these feature types. Performance of GCN is extremely good, when nucleotide occurrences with k-mer size of as 1 is given as features for the sequences in the network graph. Providing the position of nucleotide in a sequence as a feature for nodes in the graph is important in off-target prediction and GCN can predict with high auROC values.
- The off-target dataset is heavily imbalanced and cluster data sampling is done to overcome this imbalance issue by randomly sampling the majority class, NOT sequences with respect to the count of OT sequences in each sgRNA cluster.
- StellarGraph, a user friendly API, can be used to create network graphs and perform validation using a GNN model, if the off-target dataset is not highly imbalanced, as this API provides many in-built codes to enable researchers to automate the creation and normalization of adjacency and feature matrices without much manual effort.
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Terns, M.P.; Terns, R.M. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 2011, 14, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef]
- Zhang, Y.; Ge, X.; Yang, F.; Zhang, L.; Zheng, J.; Tan, X.; Jin, Z.B.; Qu, J.; Gu, F. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 2014, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Kimura, P.; Nakane, T.; Ishitani, R.; Hatada, I.; Zhang, F.; Nishimasu, H.; Nureki, O.; Slaymaker, M.; Li, Y.; Fedorova, I.; et al. Molecular mechanism of CRISPR. Found. Crystallogr. 2014, 156, 935–949. [Google Scholar]
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.; Li, Z.; Peterson, R.T.; Yeh, J.R.J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485. [Google Scholar] [CrossRef] [Green Version]
- Chiarle, R.; Zhang, Y.; Frock, R.L.; Lewis, S.M.; Molinie, B.; Ho, Y.J.; Myers, D.R.; Choi, V.W.; Compagno, M.; Malkin, D.J.; et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 2011, 147, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, S.; Kim, S.; Park, J.; Kim, J.S. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 2016, 26, 406–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.I.; Kim, J.S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wu, X.; Wang, J.; Wang, Y.; Qiu, Z.; Chang, T.; Huang, H.; Lin, R.J.; Yee, J.K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015, 33, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.J.; Webber, B.R.; Knipping, F.; Lonetree, C.l.; Tennis, N.; DeFeo, A.P.; McElroy, A.N.; Starker, C.G.; Lee, C.; Merkel, S.; et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol. Ther. 2016, 24, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kuscu, C.; Quinlan, A.; Qi, Y.; Adli, M. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015, 43, e118. [Google Scholar] [CrossRef]
- Stemmer, M.; Thumberger, T.; del Sol Keyer, M.; Wittbrodt, J.; Mateo, J.L. CCTop: An intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE 2015, 10, e0124633. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Listgarten, J.; Weinstein, M.; Kleinstiver, B.P.; Sousa, A.A.; Joung, J.K.; Crawford, J.; Gao, K.; Hoang, L.; Elibol, M.; Doench, J.G.; et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2018, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüsi, I.; Junior, J.C.J.; Gorbova, J.; Baró, X.; Escalera, S.; Demirel, H.; Allik, J.; Ozcinar, C.; Anbarjafari, G. Joint challenge on dominant and complementary emotion recognition using micro emotion features and head-pose estimation: Databases. In Proceedings of the 2017 12th IEEE International Conference on Automatic Face & Gesture Recognition (FG 2017), Washington, DC, USA, 30 May–3 June 2017; pp. 809–813. [Google Scholar]
- Domnich, A.; Anbarjafari, G. Responsible AI: Gender bias assessment in emotion recognition. arXiv 2021, arXiv:2103.11436. [Google Scholar]
- Tammvee, M.; Anbarjafari, G. Human activity recognition-based path planning for autonomous vehicles. Signal Image Video Process. 2020, 1–8. [Google Scholar] [CrossRef]
- Karabulut, D.; Tertychnyi, P.; Arslan, H.S.; Ozcinar, C.; Nasrollahi, K.; Valls, J.; Vilaseca, J.; Moeslund, T.B.; Anbarjafari, G. Cycle-consistent generative adversarial neural networks based low quality fingerprint enhancement. Multimed. Tools Appl. 2020, 1–21. [Google Scholar] [CrossRef]
- Avots, E.; Jermakovs, K.; Bachmann, M.; Paeske, L.; Ozcinar, C.; Anbarjafari, G. Ensemble approach for detection of depression using EEG features. arXiv 2021, arXiv:2103.08467. [Google Scholar]
- Novichkova, S.; Egorov, S.; Daraselia, N. MedScan, a natural language processing engine for MEDLINE abstracts. Bioinformatics 2003, 19, 1699–1706. [Google Scholar] [CrossRef] [Green Version]
- Arslan, H.S.; Sirts, K.; Fishel, M.; Anbarjafari, G. Multimodal sequential fashion attribute prediction. Information 2019, 10, 308. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.N.; Yan, X.; You, Z.H.; Zhou, X.; Li, L.P.; Wang, L.; Song, K.J. NLPEI: A Novel Self-Interacting Protein Prediction Model Based on Natural Language Processing and Evolutionary Information. Evol. Bioinform. 2020, 16, 1176934320984171. [Google Scholar] [CrossRef]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 80. [Google Scholar] [CrossRef]
- Lin, J.; Wong, K.C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics 2018, 34, i656–i663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; He, D.; Xie, L. Prediction of off-target specificity and cell-specific fitness of CRISPR-Cas System using attention boosted deep learning and network-based gene feature. PLoS Comput. Biol. 2019, 15, e1007480. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, X.; Liu, G.; Li, B.; Liu, X. Deep learning improves the ability of sgRNA off-target propensity prediction. BMC Bioinform. 2020, 21, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, X.; Cheng, L.; Luo, Y. An overview and metanalysis of machine and deep learning-based CRISPR gRNA design tools. RNA Biol. 2020, 17, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J. Prediction of sgRNA on-target activity in bacteria by deep learning. BMC Bioinform. 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutrouli, M.; Karatzas, E.; Paez-Espino, D.; Pavlopoulos, G.A. A guide to conquer the biological network era using graph theory. Front. Bioeng. Biotechnol. 2020, 8, 34. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, G.; Zhang, Z.; Yang, C.; Liu, Z.; Wang, L.; Li, C.; Sun, M. Graph neural networks: A review of methods and applications. arXiv 2018, arXiv:1812.08434. [Google Scholar]
- Kawamoto, T.; Tsubaki, M.; Obuchi, T. Mean-field theory of graph neural networks in graph partitioning. J. Stat. Mech. Theory Exp. 2019, 2019, 124007. [Google Scholar] [CrossRef] [Green Version]
- Data61, C. StellarGraph Machine Learning Library. Available online: https://github.com/stellargraph/stellargraph (accessed on 14 May 2021).
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Muhammad Rafid, A.H.; Toufikuzzaman, M.; Rahman, M.S.; Rahman, M.S. CRISPRpred (SEQ): A sequence-based method for sgRNA on target activity prediction using traditional machine learning. BMC Bioinform. 2020, 21, 1–13. [Google Scholar] [CrossRef]
- Xue, L.; Tang, B.; Chen, W.; Luo, J. Prediction of CRISPR sgRNA activity using a deep convolutional neural network. J. Chem. Inf. Model. 2018, 59, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.; Swart, P.; S Chult, D. Exploring Network Structure, Dynamics, and Function Using NetworkX; Technical Report; Los Alamos National Lab. (LANL): Los Alamos, NM, USA, 2008. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster Data Samples | sgRNA | OT | NOT |
|---|---|---|---|
| Imbalanced_OT Clusters (OT > NOT) | 29 | 626 | 304 |
| Balanced Clusters (OT = NOT) | 29 | 626 | 626 |
| Imbalanced_NOT Clusters (OT < NOT) | 29 | 626 | 1252 |
| Feature Types | Balanced Clusters | Imbalanced_NOT Clusters | Imbalanced_OT Clusters |
|---|---|---|---|
| Nucleotide Occurrence (k = 1) | 0.954 | 0.987 | 0.976 |
| Nucleotide Occurrence (k = 2) | 0.889 | 0.914 | 0.931 |
| Nucleotide Occurrence (k = 3) | 0.888 | 0.888 | 0.893 |
| Nucleotide Position | 0.925 | 0.884 | 0.879 |
| Model | auROC Value |
|---|---|
| DeepCrispr | 0.857 |
| AttnToMismatch_CNN | 0.970 |
| CnnCrispr | 0.984 |
| GCN-CRISPR | 0.987 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinodkumar, P.K.; Ozcinar, C.; Anbarjafari, G. Prediction of sgRNA Off-Target Activity in CRISPR/Cas9 Gene Editing Using Graph Convolution Network. Entropy 2021, 23, 608. https://doi.org/10.3390/e23050608
Vinodkumar PK, Ozcinar C, Anbarjafari G. Prediction of sgRNA Off-Target Activity in CRISPR/Cas9 Gene Editing Using Graph Convolution Network. Entropy. 2021; 23(5):608. https://doi.org/10.3390/e23050608
Chicago/Turabian StyleVinodkumar, Prasoon Kumar, Cagri Ozcinar, and Gholamreza Anbarjafari. 2021. "Prediction of sgRNA Off-Target Activity in CRISPR/Cas9 Gene Editing Using Graph Convolution Network" Entropy 23, no. 5: 608. https://doi.org/10.3390/e23050608