
Density of Avoided Crossings and Diabatic Representation



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Singularities in the Spectra of Random Matrices
2.1. Parameter-Dependent Random Matrices
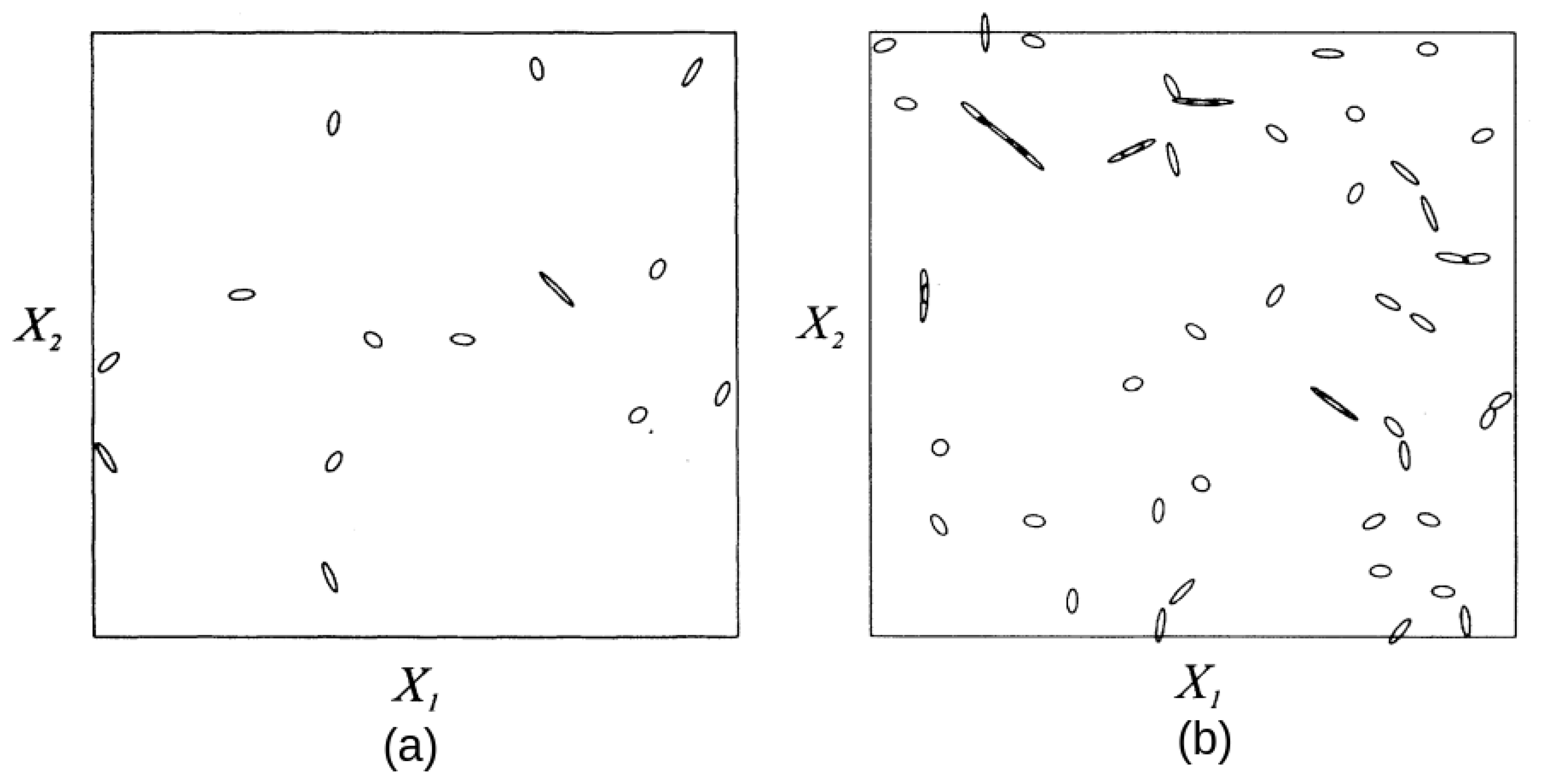
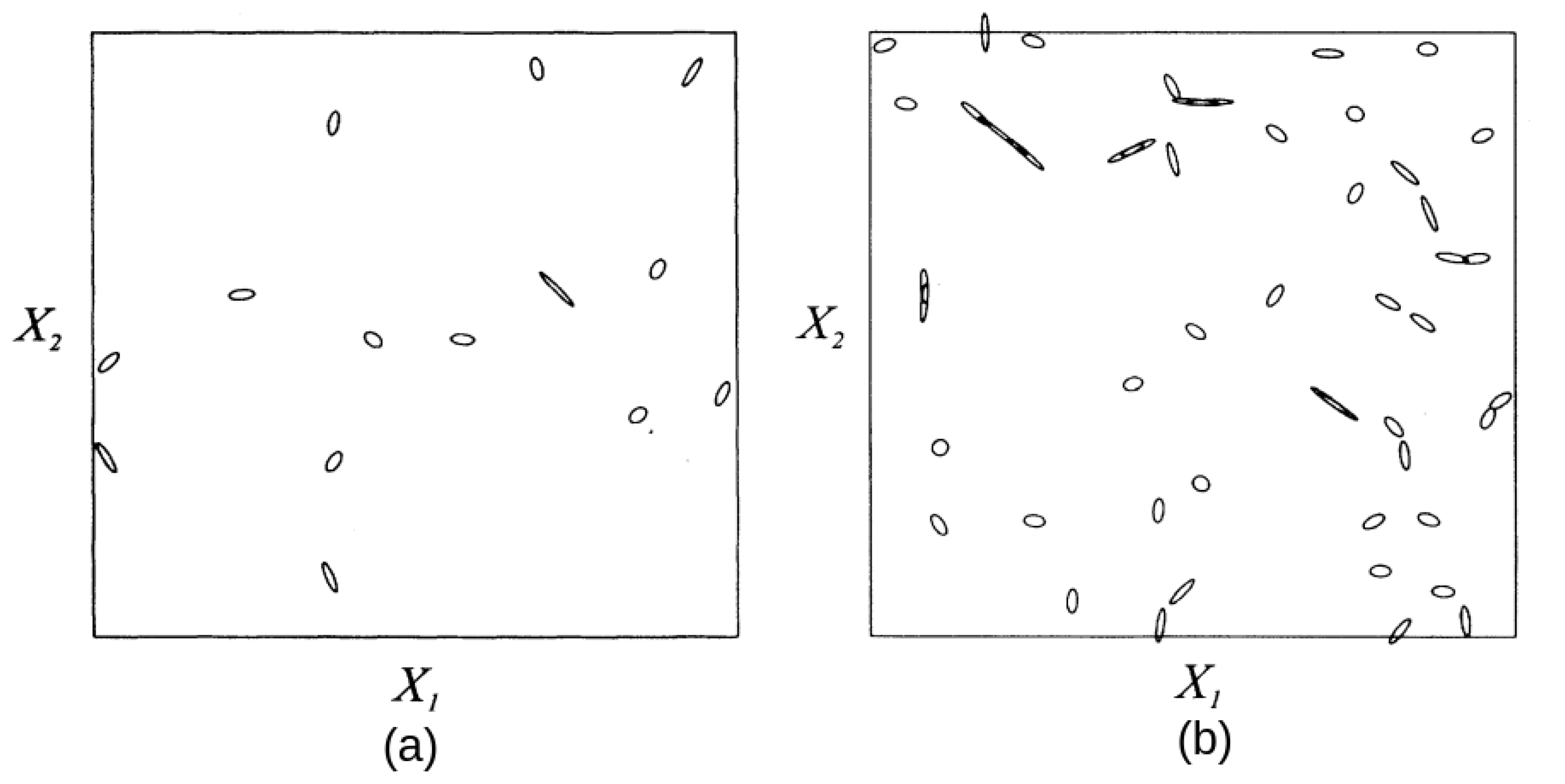
2.2. Geometrical Properties of Conical Intersections
2.3. Geometrical Properties of Avoided Crossings
3. Thermal Fluctuations in Solids
3.1. Born–Oppenheimer Approximation
3.2. Supercell Technique
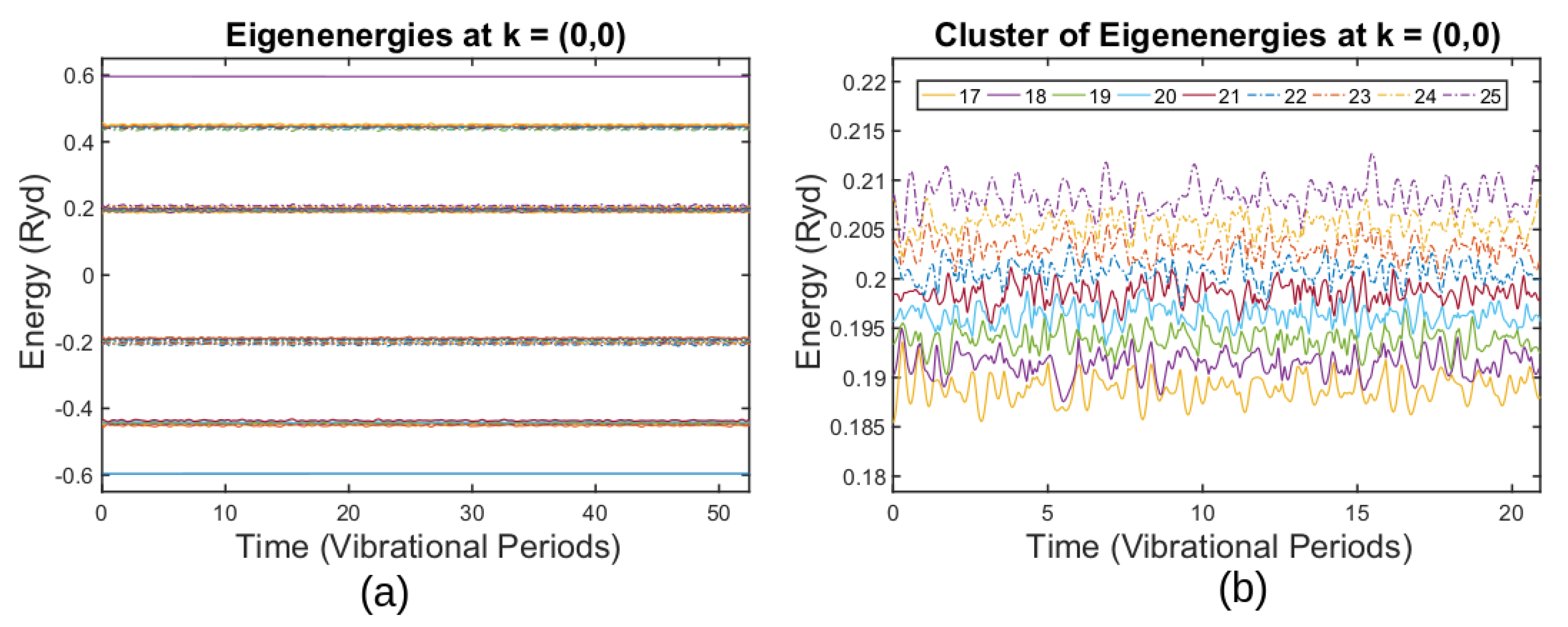
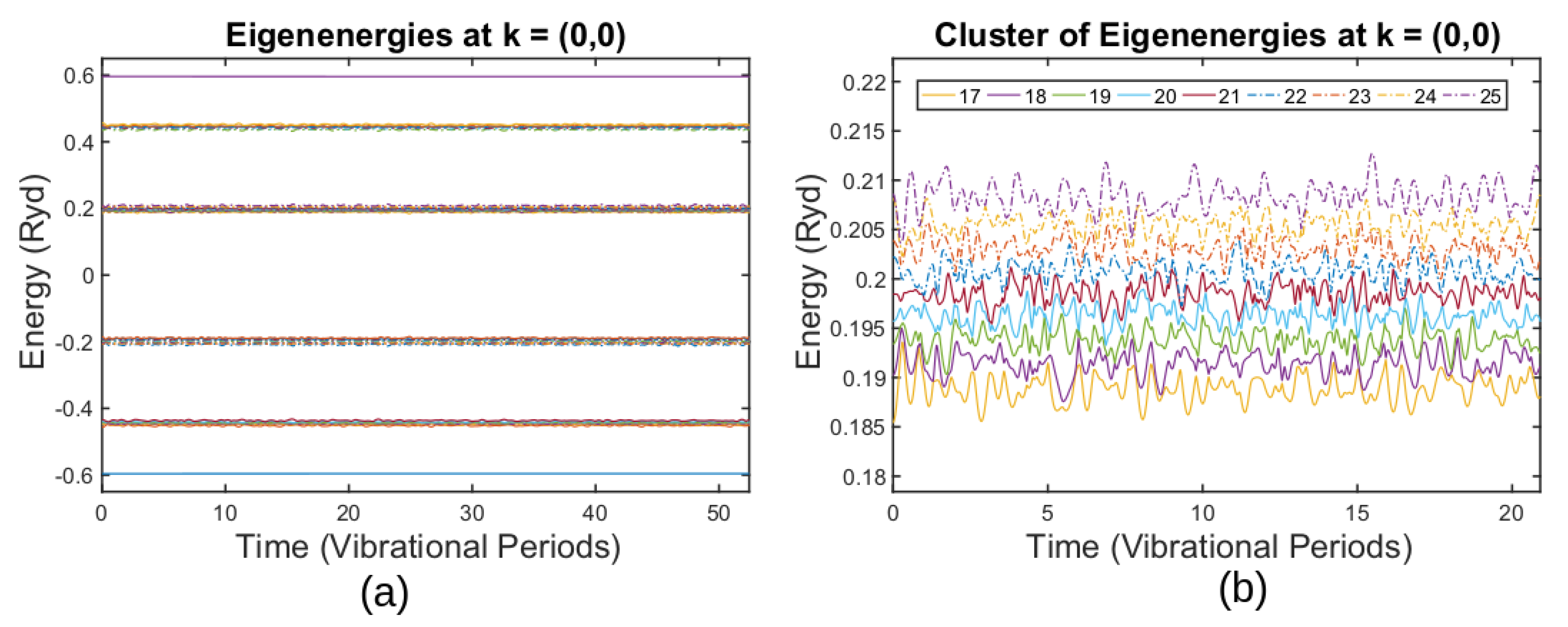
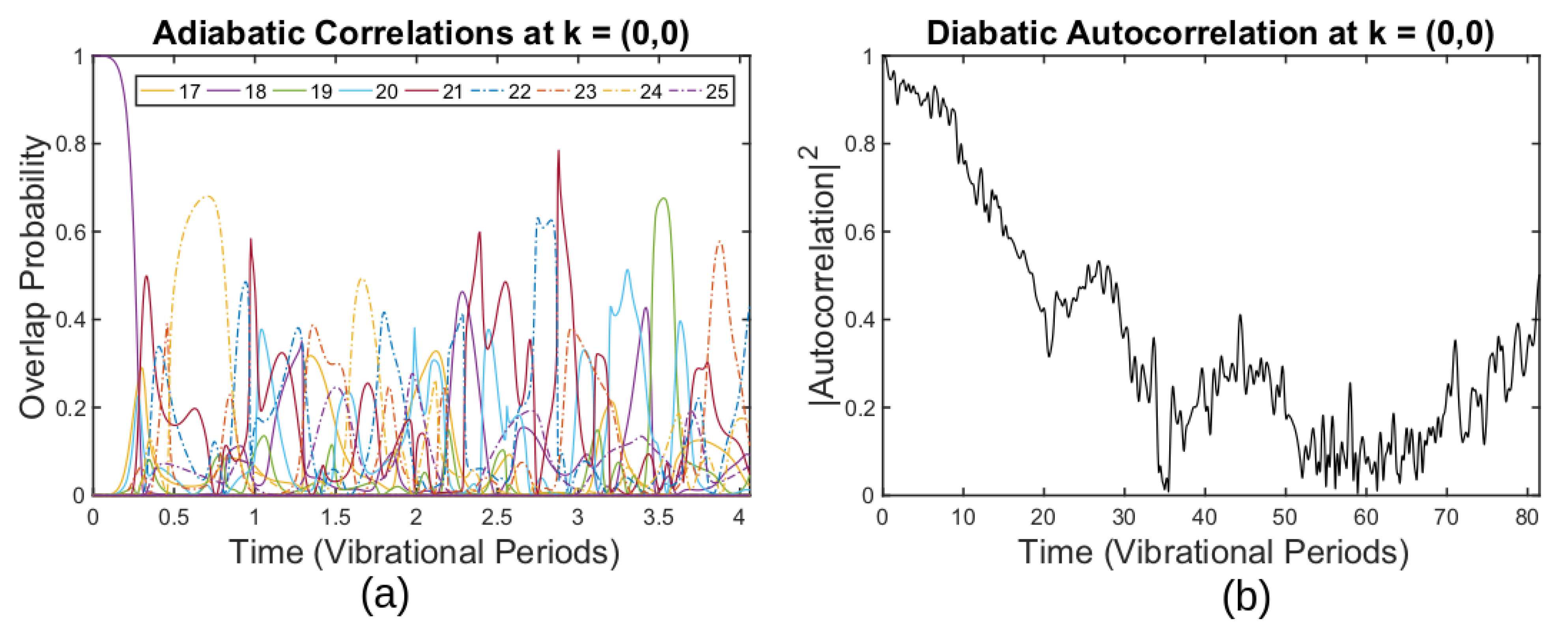
3.3. Simulating Graphene Thermal Fluctuations
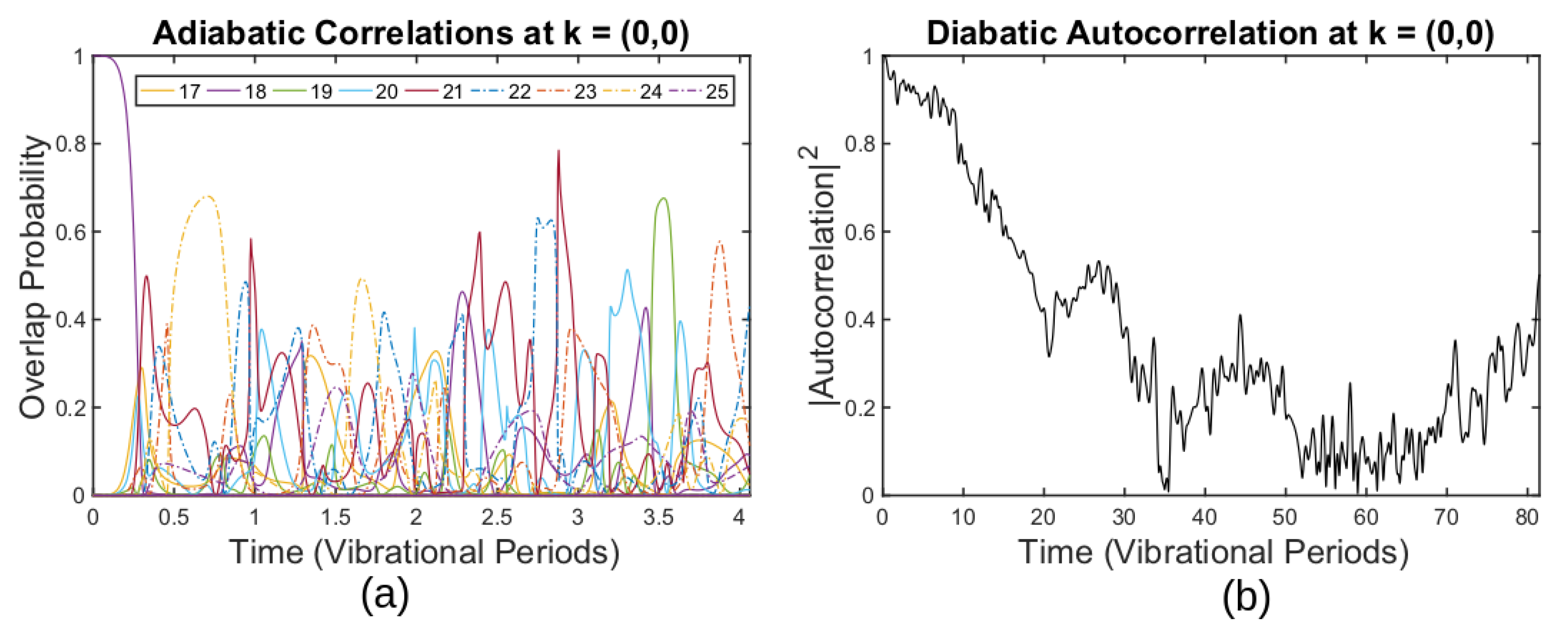
3.4. Breakdown of the ABO for Thermally Fluctuating Graphene
4. Discussion and Further Work
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABO | Adiabatic Born–Oppenheimer approximation |
| DBO | Diabatic Born–Oppenheimer approximation |
| GOE | Gaussian orthogonal ensemble |
| GUE | Gaussian unitary ensemble |
| GSE | Gaussian symplectic ensemble |
References
- Donghwan, K.; Aydin, A.; Daza, A.; Avanaki, K.N.; Keski-Rahkonen, J.; Heller, E.J. Coherent charge carrier dynamics in the presence of thermal lattice vibrations. Phys. Rev. B 2022, 106, 054311. [Google Scholar]
- Mohanty, V.; Heller, E.J. Lazy electrons in graphene. Proc. Natl. Acad. Sci. USA 2019, 116, 18316–18321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haake, F. Quantum Signatures of Chaos; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Scharf, R.; Dietz, B.; Kuś, M.; Haake, F.; Berry, M.V. Kramers’ Degeneracy and Quartic Level Repulsion. Europhys. Lett. 1988, 5, 383–389. [Google Scholar] [CrossRef]
- Grobe, R.; Haake, F.; Sommers, H.-J. Quantum Distinction of Regular and Chaotic Dissipative Motion. Phys. Rev. Lett. 1988, 61, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M. Statistical aspects of dissipation by Landau-Zener transitions. J. Phys. A Math. Gen. 1988, 21, 4021–4037. [Google Scholar] [CrossRef]
- Wilkinson, M.; Austin, E.J. Suppression of dissipation by localization. J. Phys. A Math. Gen. 1990, 23, L957–L963. [Google Scholar] [CrossRef]
- Lenz, G.; Haake, F. Transitions between universality classes of random matrices. Phys. Rev. Lett. 1990, 65, 2325–2328. [Google Scholar] [CrossRef]
- Lenz, G.; Haake, F. Reliability of small matrices for large spectra with nonuniversal fluctuations. Phys. Rev. Lett. 1991, 67, 1. [Google Scholar] [CrossRef]
- Lenz, G.; Haake, F. Classical Hamiltonian Dynamics of Rescaled Quantum Levels. Europhys. Lett. 1990, 13, 577–582. [Google Scholar]
- Wilkinson, M.; Austin, E.J. Densities of degeneracies and near-degeneracies. Phys. Rev. A 1993, 47, 2601–2609. [Google Scholar] [CrossRef]
- Wilkinson, M. A semiclassical sum rule for matrix elements of classically chaotic systems. J. Phys. A 1987, 20, 2415–2423. [Google Scholar] [CrossRef]
- Pandey, A. Statistical properties of many-particle spectra: III. Ergodic behavior in random-matrix ensembles. Ann. Phys. 1979, 119, 170–191. [Google Scholar] [CrossRef]
- Teller, E. The Crossing of Potential Surfaces. J. Phys. Chem. 1937, 41, 109–116. [Google Scholar] [CrossRef]
- Longuett-Higgins, H.C. The intersection of potential energy surfaces in polyatomic molecules. Proc. R. Soc. Lond. A 1975, 344, 147–156. [Google Scholar]
- Berry, M.V. Quantal phase factors accompanying adiabatic changes. Proc. R. Soc. Lond. A 1984, 392, 45–57. [Google Scholar]
- Selberg, A. Remarks on a multiple integral. Nor. Mat. Tidsskr. 1944, 26, 71–78. [Google Scholar]
- Aomoto, K. Jacobi Polynomials Associated with Selberg Integrals. SIAM J. Math. Anal. 1987, 18, 545–549. [Google Scholar] [CrossRef]
- Mehta, M.L. Random Matrices; Academic Press: New York, NY, USA, 1990. [Google Scholar]
- Walker, P.N.; Sanchez, M.J.; Wilkinson, M. ingularities in the Spectra of Random Matrices. J. Math. Phys. 1996, 37, 5019–5032. [Google Scholar] [CrossRef]
- Berry, M.V.; Wilkinson, M. Diabolical points in the spectra of triangles. Proc. R. Soc. Lond. A 1984, 392, 15–43. [Google Scholar]
- Walker, P.N.; Wilkinson, M. Universal Fluctuations of Chern Integers. Phys. Rev. Lett. 1995, 74, 4055–4058. [Google Scholar] [CrossRef]
- Kuś, M.; Scharf, R.; Haake, F. Symmetry versus degree of level repulsion for kicked quantum systems. Z. Phys. B 1987, 66, 129–134. [Google Scholar] [CrossRef]
- Haake, F.; Kuś, M.; Scharf, R. Classical and Quantum Chaos for a Kicked Top. Z. Phys. B 1987, 65, 381–395. [Google Scholar] [CrossRef]
- Zakrzewski, J.; Delande, D. Parametric motion of energy levels in quantum chaotic systems. I. Curvature distributions. Phys. Rev. E 1993, 47, 1650–1664. [Google Scholar] [CrossRef] [PubMed]
- von Oppen, F. Exact distribution of eigenvalue curvatures of chaotic quantum systems. Phys. Rev. Lett. 1994, 73, 798–801. [Google Scholar] [CrossRef] [PubMed]
- von Neumann, J.; Wigner, E. Uber merkwürdige diskrete Eigenwerte. Uber das Verhalten von Eigenwerten bei adiabatischen Prozessen. Phys. Z. 1929, 30, 467–470. [Google Scholar]
- Austin, E.J.; Wilkinson, M. Statistical properties of parameter-dependent classically chaotic quantum systems. Nonlinearity 1992, 5, 1137–1150. [Google Scholar] [CrossRef]
- Zakrzewski, J.; Kuś, M. Distributions of avoided crossings for quantum chaotic systems. Phys. Rev. Lett. 1991, 67, 2749–2752. [Google Scholar] [CrossRef]
- Zakrzewski, J.; Delande, D.; Kuś, M. Parametric motion of energy levels in quantum chaotic systems. II. Avoided-crossing distributions. Phys. Rev. E 1993, 47, 1665–1676. [Google Scholar] [CrossRef]
- Grobe, R.; Haake, F. Universality of cubic-level repulsion for dissipative quantum chaos. Phys. Rev. Lett. 1989, 62, 2893–2896. [Google Scholar] [CrossRef]
- Born, M.; Oppenheimer, R. Zur quantentheorie der Molekeln. Ann. Phys. 1927, 389, 457–484. [Google Scholar] [CrossRef]
- Heller, E.J. The Semiclassical Way to Dynamics and Spectroscopy; Princeton University Press: Princeton, NJ, USA, 2018. [Google Scholar]
- Born, M. Über die Serienspektra der Elemente. Z. Phys. 1920, 2, 423–469. [Google Scholar]
- Heller, E.J.; Kim, D. Schrödinger correspondence applied to crystals. J. Phys. Chem. A 2019, 123, 4379–4388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggenthaler, M.; Tancogne-Dejean, N.; Flick, J.; Appel, H.; Rubio, A. From a quantum-electrodynamical light–matter description to novel spectroscopies. Nat. Rev. Chem. 2018, 2, 0118. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045–1097. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R. Solids and Surfaces: A Chemist’s View of Bonding in Extended Structures; VCH: New York, NY, USA, 1988. [Google Scholar]
- Turchi, P.E.A.; Gonis, A.; Colombo, L. Tight-Binding Approach to Computational Materials Science; Materials Research Society: Warrendale, PA, USA, 1998. [Google Scholar]
- Makov, G.; Payne, M.C. Periodic boundary conditions in Ab Initio Calculations. Phys. Rev. B 1995, 51, 4014–4022. [Google Scholar] [CrossRef] [Green Version]
- Makov, G.; Shah, R.; Payne, M.C. Periodic boundary conditions in ab initio calculations. II. Brillouin-zone sampling for aperiodic systems. Phys. Rev. B 1996, 53, 15513–15517. [Google Scholar] [CrossRef]
- Mizokami, K.; Togo, A.; Tanaka, I. Lattice thermal conductivities of two SiO2 polymorphs by first-principles calculations and the phonon Boltzmann transport equation. Phys. Rev. B 2018, 97, 224306. [Google Scholar] [CrossRef] [Green Version]
- Landau, L.D. On the Theory of Transfer of Energy at Collisions II. Phys. Z. Sowjetunion 1932, 2, 46–51. [Google Scholar]
- Brundobler, S.; Elser, V. S-matrix for generalized Landau-Zener problem. J. Phys. A Math. Gen. 1993, 26, 1211–1227. [Google Scholar] [CrossRef]
- Sinitsyn, N.A. Counterintuitive transitions in the multistate Landau–Zener problem with linear level crossings. J. Phys. A Math. Gen. 2004, 37, 10691–10697. [Google Scholar] [CrossRef] [Green Version]
- Sinitsyn, N.A.; Chernyak, V.Y. The quest for solvable multistate Landau-Zener models. J. Phys. A Math. Theor. 2017, 50, 255203. [Google Scholar] [CrossRef] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obzhirov, A.E.; Heller, E.J. Density of Avoided Crossings and Diabatic Representation. Entropy 2023, 25, 751. https://doi.org/10.3390/e25050751
Obzhirov AE, Heller EJ. Density of Avoided Crossings and Diabatic Representation. Entropy. 2023; 25(5):751. https://doi.org/10.3390/e25050751
Chicago/Turabian StyleObzhirov, Anatoly E., and Eric J. Heller. 2023. "Density of Avoided Crossings and Diabatic Representation" Entropy 25, no. 5: 751. https://doi.org/10.3390/e25050751
APA StyleObzhirov, A. E., & Heller, E. J. (2023). Density of Avoided Crossings and Diabatic Representation. Entropy, 25(5), 751. https://doi.org/10.3390/e25050751