
Exploring the Role of Genetic and Environmental Features in Colorectal Cancer Development: An Agent-Based Approach




Abstract
:1. Introduction
2. Methods
2.1. Biological Background
2.2. The Agent-Based Model
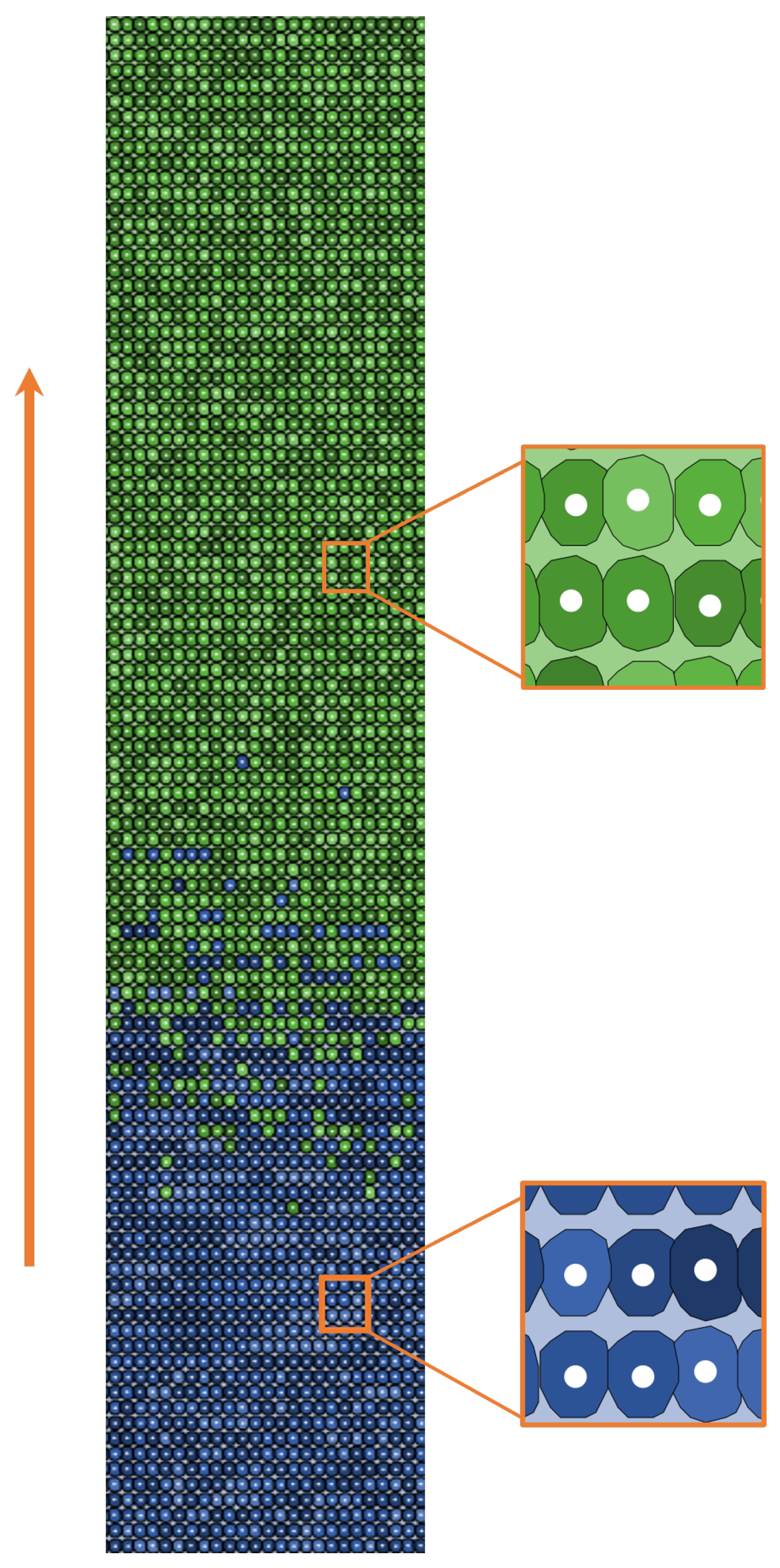
2.2.1. World Structure
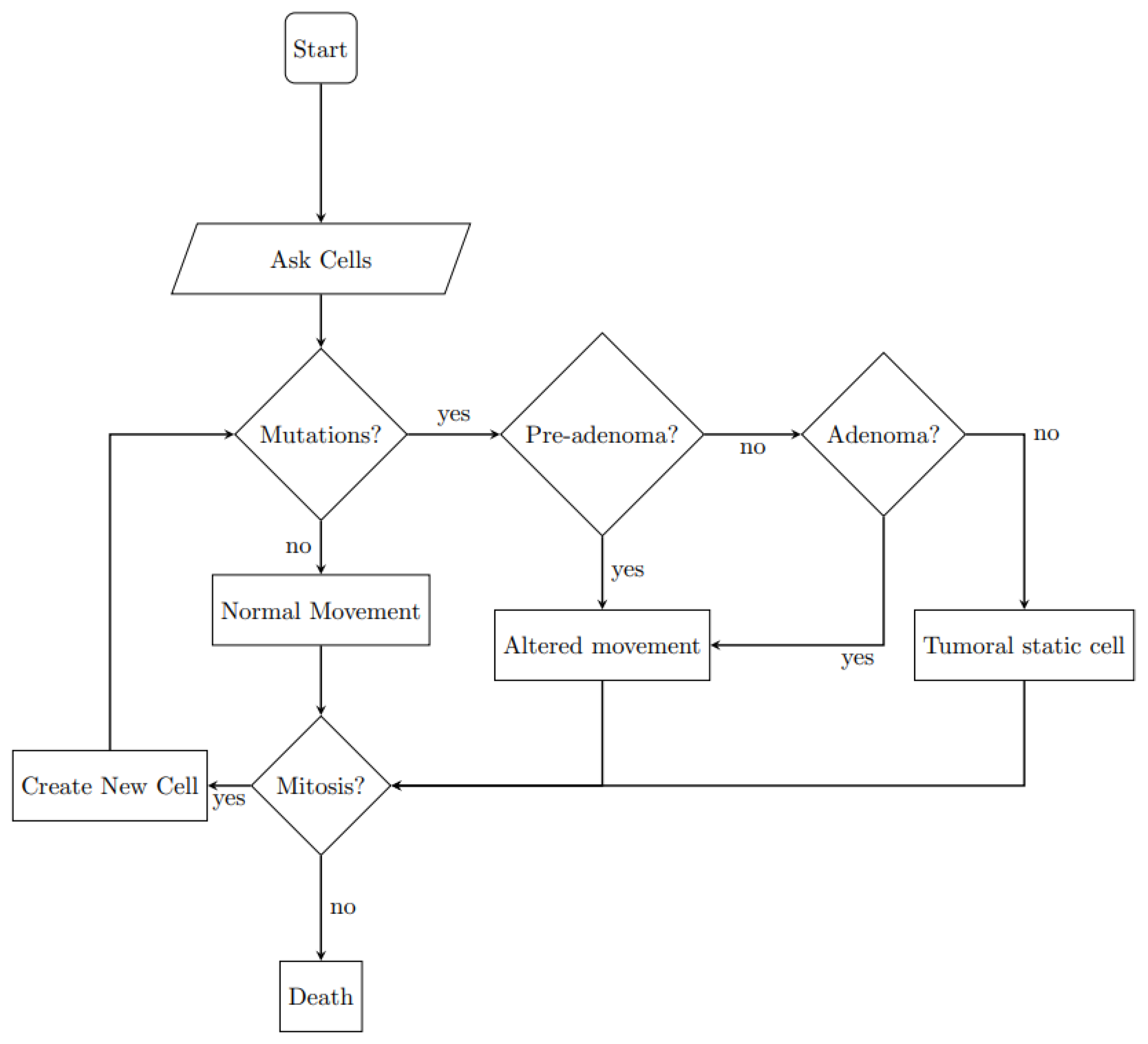
2.2.2. Agents Behavior
2.2.3. Genetic Features and Mutation Effects
2.2.4. Micro-Environmental Features
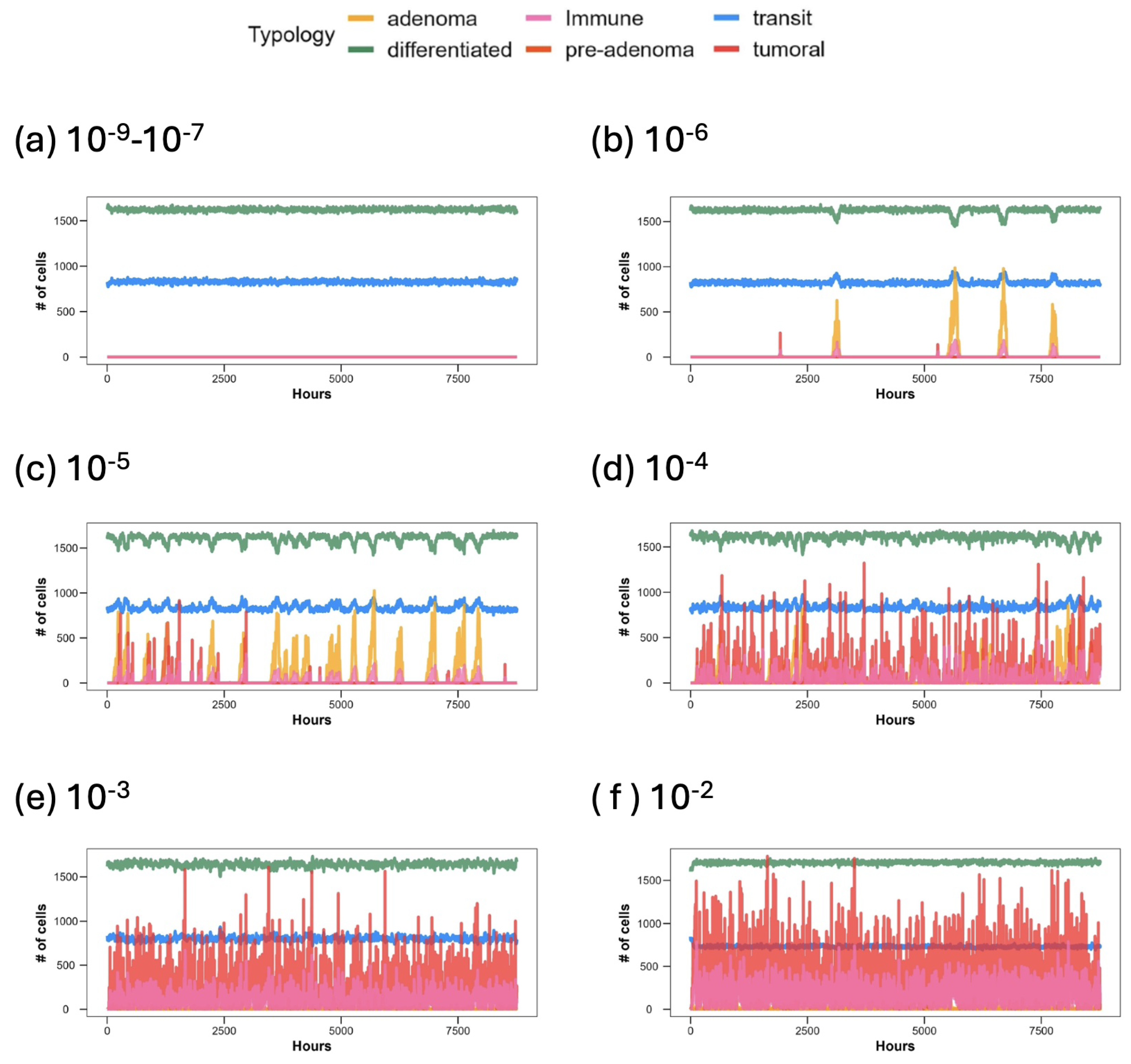
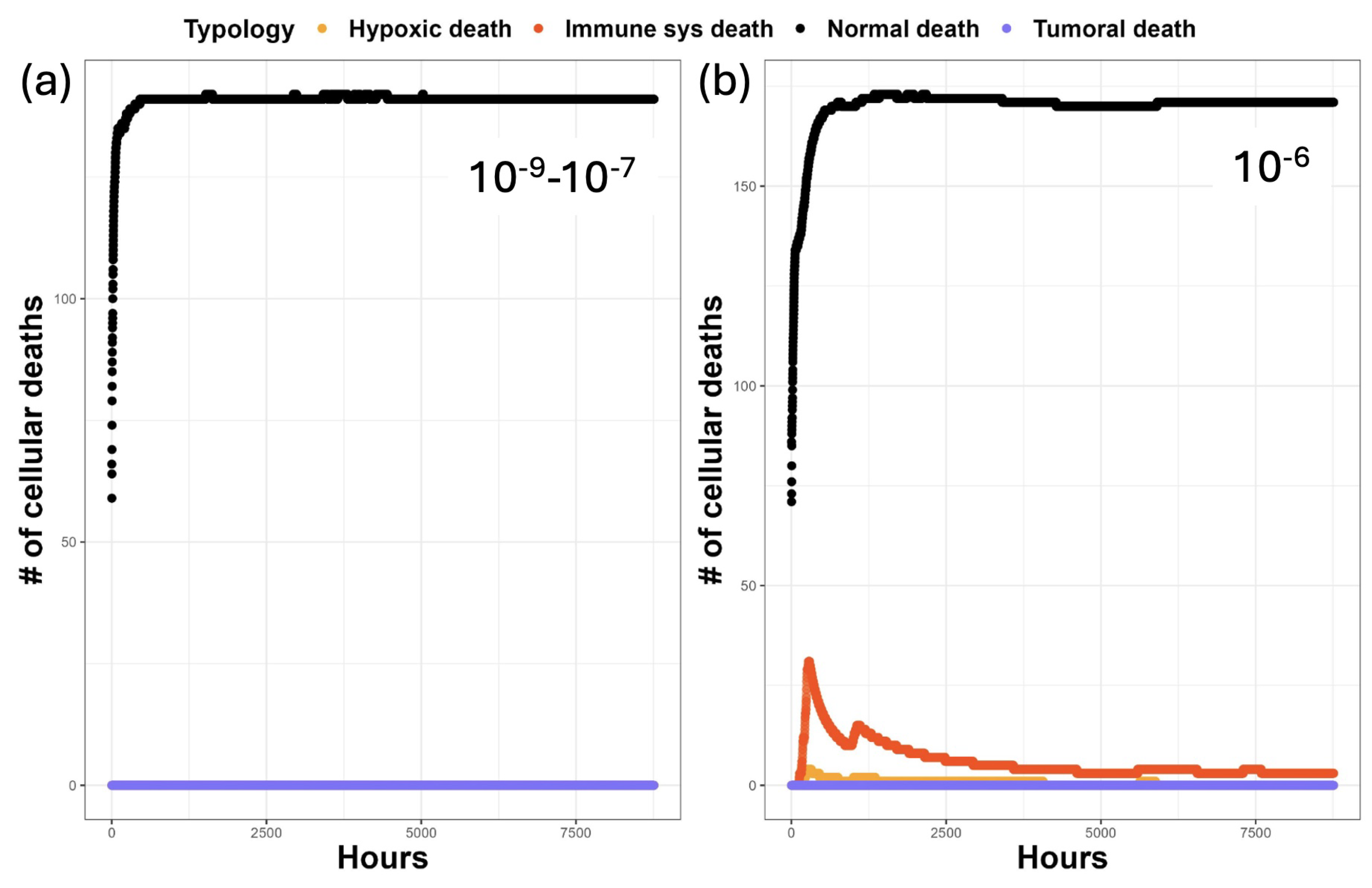
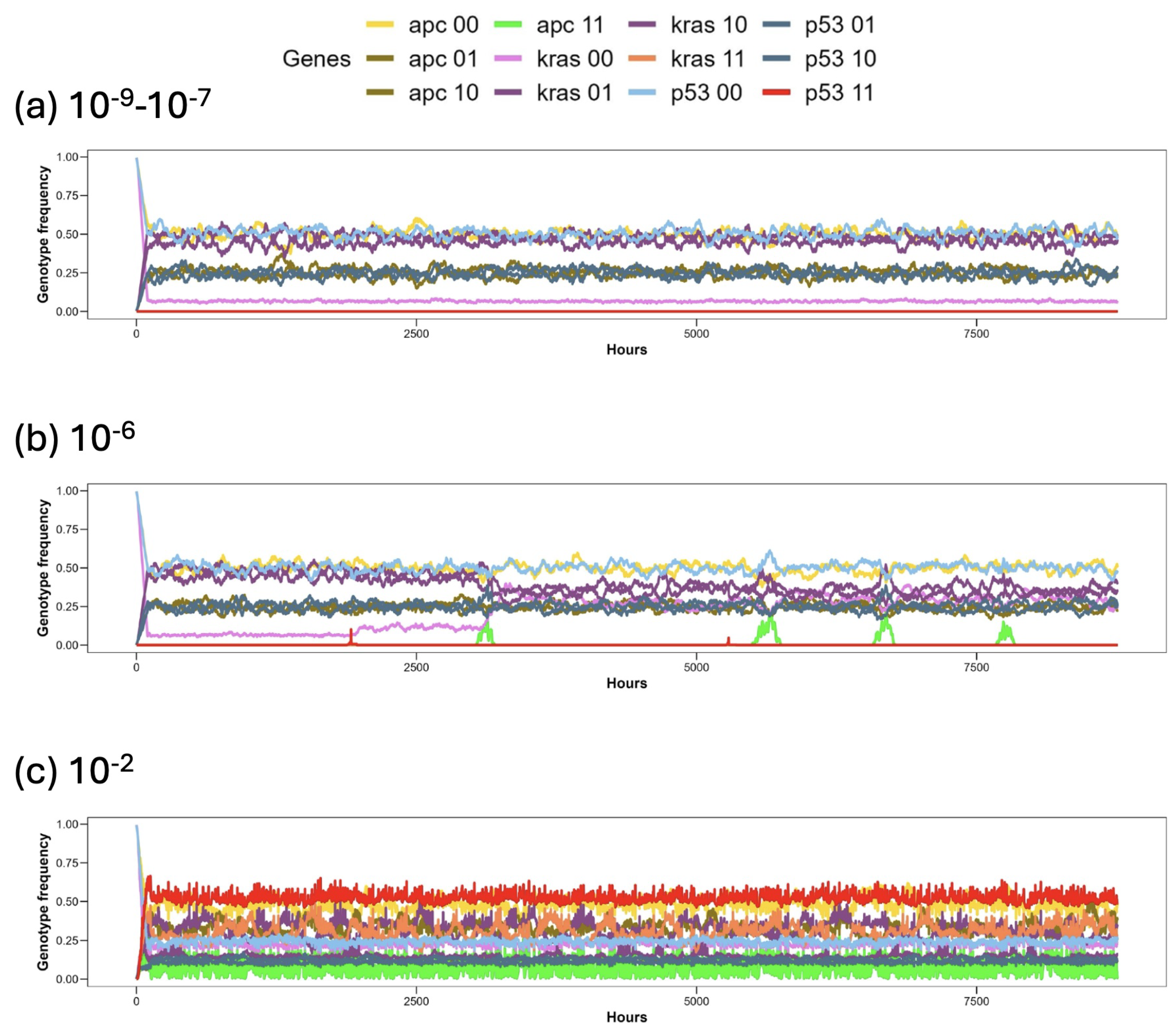
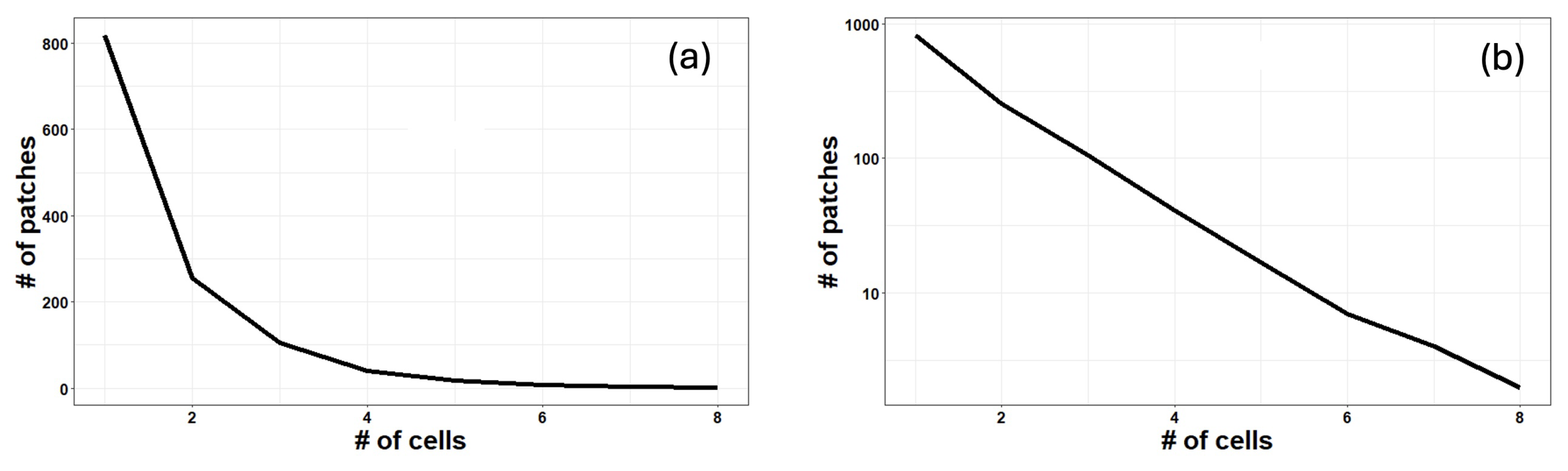
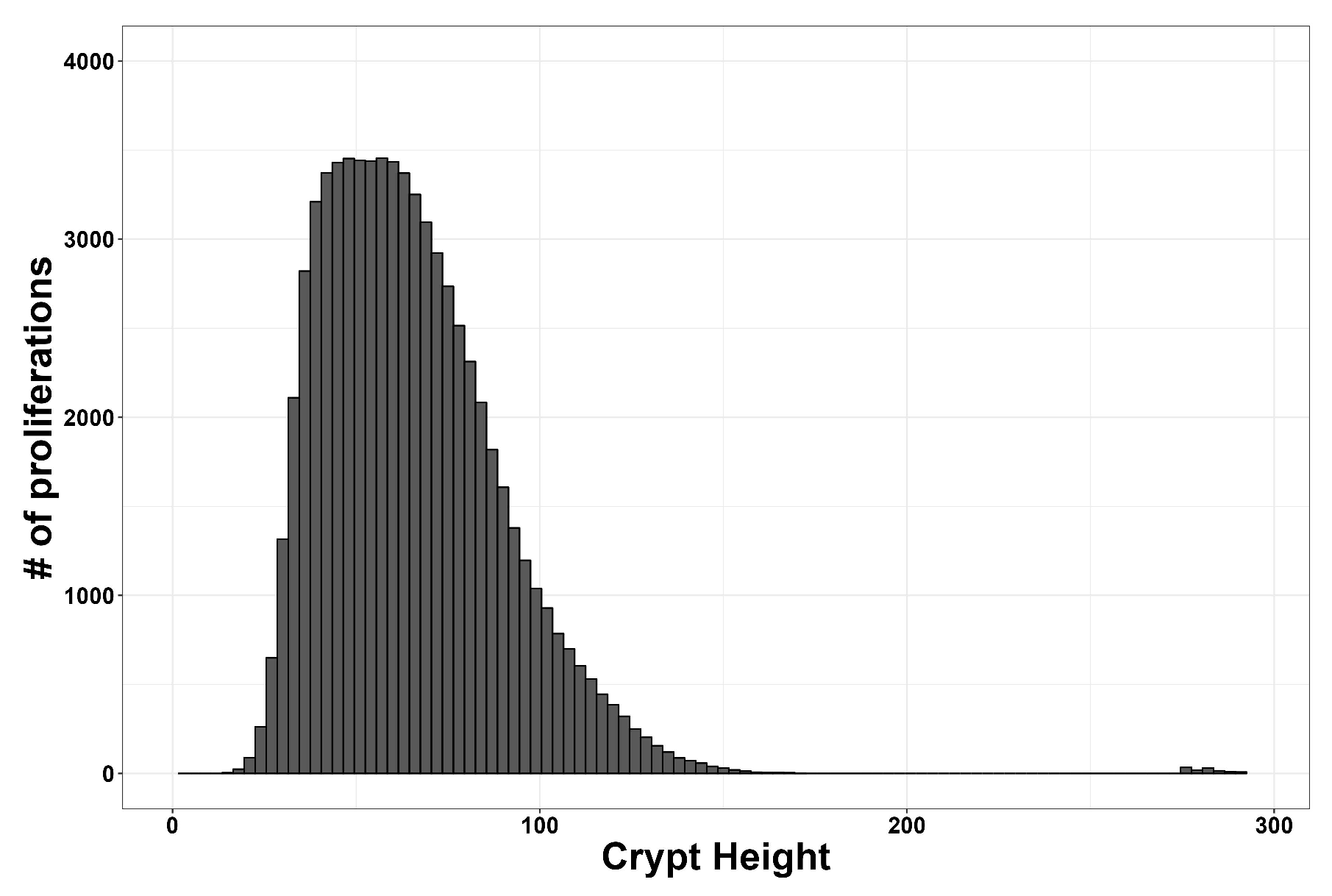
3. Results
- Number of immune system cells: 1/h
- Number of cells that a killer cell eliminates before death: 5 cells.
- Hypoxic threshold: 5 cells/patch.
- Immune system threshold: 5 cells/patch.
- Probability of starting genotype: p = 0.5.
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CRC | colorectal cancer |
| SMT | somatic mutation theory |
| TOFT | tissue organization field theory |
| TME | tumor microenvironment |
References
- Plutynski, A. How is cancer complex? Eur. J. Philos. Sci. 2021, 11, 1–30. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Biology of Cancer; Garland Science: New York City, NY, USA, 2013. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Duong, H.-Q. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef]
- Bürtin, F.; Mullins, C.S.; Linnebacher, M. Mouse models of colorectal cancer: Past, present and future perspectives. World J. Gastroenterol. 2020, 26, 1394. [Google Scholar] [CrossRef]
- Williams, A.C.; Hague, A.; Manning, A.M.; Van der Stappen, J.W.; Paraskeva, C. In vitro models of human colorectal cancer. Cancer Surveys 1993, 16, 15–29. [Google Scholar]
- Chen, H.; Cheng, Y.; Wang, X.; Wang, J.; Shi, X.; Li, X.; Tan, W.; Tan, Z. 3D printed in vitro tumor tissue model of colorectal cancer. Theranostics 2020, 10, 12127. [Google Scholar] [CrossRef]
- Johnston, M.D.; Edwards, C.M.; Bodmer, W.F.; Maini, P.K.; Chapman, S.J. Mathematical modeling of cell population dynamics in the colonic crypt and in colorectal cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 4008–4013. [Google Scholar] [CrossRef]
- Michor, F.; Iwasa, Y.; Lengauer, C.; Nowak, M.A. Dynamics of colorectal cancer. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 15, pp. 484–493. [Google Scholar]
- Meineke, F.A.; Potten, C.S.; Loeffler, M. Cell migration and organization in the intestinal crypt using a lattice-free model. Cell Prolif. 2001, 34, 253–266. [Google Scholar] [CrossRef]
- Haupt, S.; Gleim, N.; Ahadova, A.; Bläker, H.; von Knebel Doeberitz, M.; Kloor, M.; Heuveline, V. A computational model for investigating the evolution of colonic crypts during Lynch syndrome carcinogenesis. Comput. Syst. Oncol. 2021, 1, e1020. [Google Scholar] [CrossRef]
- Van Leeuwen, I.M.M.; Mirams, G.R.; Walter, A.; Fletcher, A.; Murray, P.; Osborne, J.; Varma, S.; Young, S.J.; Cooper, J.; Doyle, B.; et al. An integrative computational model for intestinal tissue renewal. Cell Prolif. 2009, 42, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Quirke, P. Experimental models of colorectal cancer. Dis. Colon Rectum 1998, 41, 490–505. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.E.; Buczacki, S.J.A.; Arends, M.J.; Adams, D.J. Mouse models of colorectal cancer as preclinical models. Bioessays 2015, 37, 909–920. [Google Scholar] [CrossRef]
- Leystra, A.A.; Clapper, M.L. Gut microbiota influences experimental outcomes in mouse models of colorectal cancer. Genes 2019, 10, 900. [Google Scholar] [CrossRef]
- Sensi, F.; D’Angelo, E.; D’Aronco, S.; Molinaro, R.; Agostini, M. Preclinical three-dimensional colorectal cancer model: The next generation of in vitro drug efficacy evaluation. J. Cell. Physiol. 2019, 234, 181–191. [Google Scholar] [CrossRef]
- Metzcar, J.; Wang, Y.; Heiland, R.; Macklin, P. A review of cell-based computational modeling in cancer biology. JCO Clin. Cancer Inform. 2019, 2, 1–13. [Google Scholar] [CrossRef]
- Ingham-Dempster, T.; Walker, D.C.; Corfe, B.M. An agent-based model of anoikis in the colon crypt displays novel emergent behaviour consistent with biological observations. R. Soc. Open Sci. 2017, 4, 160858. [Google Scholar] [CrossRef]
- Pluchino, A.; Rapisarda, A.; Garofalo, C. Efficient promotion strategies in hierarchical organizations. Phys. A Stat. Mech. Its Appl. 2011, 390, 3496–3511. [Google Scholar] [CrossRef]
- Camillen, F.; Caprií, S.; Garofalo, C.; Ignaccolo, M.; Inturri, G.; Pluchino, A.; Rapisarda, A.; Tudisco, S. Multi agent simulation of pedestrian behavior in closed spatial environments. J. Artif. Soc. Soc. Simulations 2014, 17, 16. [Google Scholar]
- Le Pira, M.; Inturri, G.; Ignaccolo, M.; Pluchino, A.; Rapisarda, A. Finding shared decisions in stakeholder networks: An agent-based approach. Phys. A Stat. Mech. Its Appl. 2017, 466, 277–287. [Google Scholar] [CrossRef]
- Fichera, A.; Pluchino, A.; Volpe, R. A multi-layer agent-based model for the analysis of energy distribution networks in urban areas. Phys. A Stat. Mech. Its Appl. 2018, 508, 710–725. [Google Scholar] [CrossRef]
- Fazio, M.; Pluchino, A.; Inturri, G.; Le Pira, M.; Giuffrida, N.; Ignaccolo, M. Exploring the impact of mobility restrictions on the COVID-19 spreading through an agent-based approach. J. Transp. Health 2022, 25, 101373. [Google Scholar] [CrossRef] [PubMed]
- Conti, E.; Di Mauro, L.S.; Pluchino, A.; Mulder, C. Testing for top-down cascading effects in a biomass-driven ecological network of soil invertebrates. Ecol. Evol. 2020, 10, 7062–7072. [Google Scholar] [CrossRef]
- Zhang, B.; DeAngelis, D.L. An overview of agent-based models in plant biology and ecology. Ann. Bot. 2020, 126, 539–557. [Google Scholar] [CrossRef]
- Glen, C.M.; Kemp, M.L.; Voit, E.O. Agent-based modeling of morphogenetic systems: Advantages and challenges. PLoS Comput. Biol. 2019, 15, e1006577. [Google Scholar] [CrossRef]
- An, G.; Mi, Q.; Dutta-Moscato, J.; Vodovotz, Y. Agent-based models in translational systems biology. WIREs Syst. Biol. Med. 2009, 1, 159–171. [Google Scholar] [CrossRef]
- Ingham-Dempster, T.A.; Rosser, R.; Corfe, B.M.; Walker, D.C. From cell to multi-crypt: Agent-based models of the human colon suggests novel processes of Field cancerisation. J. Comput. Sci. 2020, 41, 101066. [Google Scholar] [CrossRef]
- Mirams, G.R.; Fletcher, A.G.; Maini, P.K.; Byrne, H.M. A theoretical investigation of the effect of proliferation and adhesion on monoclonal conversion in the colonic crypt. J. Theor. Biol. 2012, 312, 143–156. [Google Scholar] [CrossRef]
- Niida, A.; Mimori, K.; Shibata, T.; Miyano, S. Modeling colorectal cancer evolution. J. Hum. Genet. 2021, 66, 869–878. [Google Scholar] [CrossRef]
- Doyle, J.J. Cell types as species: Exploring a metaphor. Front. Plant Sci. 2022, 13, 868565. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H. What is a cell type and how to define it? Cell 2022, 185, 2739–2755. [Google Scholar] [CrossRef] [PubMed]
- Tëmkin, I.; Eldredge, N. Networks and hierarchies: Approaching complexity in evolutionary theory. In Macroevolution; Springer: Berlin/Heidelberg, Germany, 2015; pp. 183–226. [Google Scholar]
- Bernstein, C.; Facista, A.; Nguyen, H.; Zaitlin, B.; Hassounah, N.; Loustaunau, C.; Payne, C.M.; Banerjee, B.; Goldschmid, S.; Tsikitis, V.L.; et al. Cancer and age-related colonic crypt deficiencies in cytochrome c oxidase I. World J. Gastrointest. Oncol. 2010, 2, 429. [Google Scholar] [CrossRef] [PubMed]
- Humphries, A.; Wright, N.A. Colonic crypt organization and tumorigenesis. Nat. Rev. Cancer 2008, 8, 415–424. [Google Scholar] [CrossRef]
- Van Der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Wilensky, U. NetLogo. 1999. Available online: http://ccl.northwestern.edu/netlogo/ (accessed on 1 January 2021).
- Potten, C.S.; Chwalinski, S.; Swindell, R.; Palmer, M. The spatial organization of the hierarchical proliferative cells of the crypts of the small intestine into clusters of ‘synchronized’ cells. Cell Prolif. 1982, 15, 351–370. [Google Scholar] [CrossRef]
- Potten, C.S.; Kellett, M.; Rew, D.A.; Roberts, S.A. Proliferation in human gastrointestinal epithelium using bromodeoxyuridine in vivo: Data for different sites, proximity to a tumour, and polyposis coli. Gut 1992, 33, 524. [Google Scholar] [CrossRef]
- Bravo, R.; Axelrod, D.E. A calibrated agent-based computer model of stochastic cell dynamics in normal human colon crypts useful for in silico experiments. Theor. Biol. Med. Model. 2013, 10, 1–24. [Google Scholar] [CrossRef]
- Zhao, R.; Michor, F. Patterns of proliferative activity in the colonic crypt determine crypt stability and rates of somatic evolution. PLoS Comput. Biol. 2013, 9, e1003082. [Google Scholar] [CrossRef]
- Yatabe, Y.; Tavaré, S.; Shibata, D. Investigating stem cells in human colon by using methylation patterns. Proc. Natl. Acad. Sci. USA 2001, 98, 10839–10844. [Google Scholar] [CrossRef]
- Nowak, M.A. Evolutionary Dynamics: Exploring the Equations of Life; Harvard University Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Baker, A.-M.; Cereser, B.; Melton, S.; Fletcher, A.G.; Rodriguez-Justo, M.; Tadrous, P.J.; Humphries, A.; Elia, G.; McDonald, S.A.C.; Wright, N.A.; et al. Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 2014, 8, 940–947. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Stein, R.; Wichmann, H.-E.; Potten, C.S.; Kaur, P.; Chwalinski, S. Intestinal cell proliferation. I. A comprehensive model of steady-state proliferation in the crypt. Cell Prolif. 1986, 19, 627–645. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, A.; Bowling, S.; Rodriguez, T.A. Cell competition and its role in the regulation of cell fitness from development to cancer. Dev. Cell 2016, 38, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Komarova, N.L.; Sengupta, A.; Jallepalli, P.V.; Shih, I.-M.; Vogelstein, B.; Lengauer, C. The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. USA 2002, 99, 16226–16231. [Google Scholar] [CrossRef]
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Cancer 2004, 4, 197–205. [Google Scholar] [CrossRef]
- Liu, X.; Jakubowski, M.; Hunt, J.L. KRAS gene mutation in colorectal cancer is correlated with increased proliferation and spontaneous apoptosis. Am. J. Clin. Pathol. 2011, 135, 245–252. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 tumor suppressor gene: Important milestones at the various steps of tumorigenesis. Genes Cancer Sage Publ. 2011, 2, 466–474. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Guo, C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int. Immunopharmacol. 2021, 101, 108374. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Wang, Y.; Zhou, Y.; Quan, Q.; Zhang, Y.; Wang, H.; Zhang, B.; Xia, L. Immune cell concentrations among the primary tumor microenvironment in colorectal cancer patients predicted by clinicopathologic characteristics and blood indexes. J. Immunother. Cancer 2019, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sieber, O.; Heinimann, K.; Tomlinson, I. Genomic stability and tumorigenesis. Semin. Cancer Biol. 2005, 15, 61–66. [Google Scholar] [CrossRef]
- Zheng, Z.; Yu, T.; Zhao, X.; Gao, X.; Zhao, Y.; Liu, G. Intratumor heterogeneity: A new perspective on colorectal cancer research. Cancer Med. 2020, 9, 7637–7645. [Google Scholar] [CrossRef]
- Woodward, J.; Woodward, J.F. Making Things Happen: A Theory of Causal Explanation; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Woodward, J. Causation in biology: Stability, specificity, and the choice of levels of explanation. Biol. Philos. 2010, 25, 287–318. [Google Scholar] [CrossRef]
- Rondeau, E.; Larmonier, N.; Pradeu, T.; Bikfalvi, A. Characterizing causality in cancer. Elife 2019, 8, e53755. [Google Scholar] [CrossRef]
- Bechtel, W. Explicating top-down causation using networks and dynamics. Philos. Sci. 2017, 84, 253–274. [Google Scholar] [CrossRef]
- Ellis, G.F.R. Top-down causation and emergence: Some comments on mechanisms. Interface Focus 2012, 2, 126–140. [Google Scholar] [CrossRef]
- Bertolaso, M. Philosophy of Cancer; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Soto, A.M.; Sonnenschein, C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. Bioessays 2011, 33, 332–340. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Cell Variables | Values |
|---|---|
| Lifetime | physiological = min 96 h (hours), max 120 h; Tumoral = min 96 h, max 150 h. |
| Mitosis time 1 | Normal distribution, mean = Mitosis mean time, variance 1. |
| Mitosis mean time | physiological (transit-amplifying, differentiated cells) = 24 h; with KRAS heterozygote = 12 h; with typology tumoral = 10 h. |
| -cat | Descendant gradient: 1 at the bottom, 0 at the top of the crypt. |
| Neoplastic typologies | pre-adenoma: true if APC = [1 1] |
| adenoma: true if APC = [1 1] and K-ras = [1 0] or [0 1] | |
| tumoral: true if APC = [1 1], K-ras = [1 0] and P53 = [1 1] | |
| Movement | Physiological cells = ↑ |
| pre-adenoma = | |
| adenoma = | |
| tumoral = no movement | |
| Proliferation directions | Physiological cells = ⟷ |
| pre-adenoma = | |
| adenoma = | |
| tumoral = |
| Genes | State |
|---|---|
| APC | wild-type = [0 0], heterozygote = [1 0] [0 1], |
| mutated = [1 1] (trigger pre-adenoma typology) | |
| KRAS | wild-type = [0 0], heterozygote = [1 0] [0 1] |
| (trigger adenoma typology) | |
| TP53 | wild-type = [0 0], heterozygote = [1 0] [0 1], |
| mutated = [1 1] (trigger tumoral typology) | |
| Regulation genes (N = 50) | N = [ [0 0], [0 1], …, [1 0] ] |
| if a given threshold x is passed | |
| and P53 = [1 1], trigger cells death |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ledda, M.; Pluchino, A.; Ragusa, M. Exploring the Role of Genetic and Environmental Features in Colorectal Cancer Development: An Agent-Based Approach. Entropy 2024, 26, 923. https://doi.org/10.3390/e26110923
Ledda M, Pluchino A, Ragusa M. Exploring the Role of Genetic and Environmental Features in Colorectal Cancer Development: An Agent-Based Approach. Entropy. 2024; 26(11):923. https://doi.org/10.3390/e26110923
Chicago/Turabian StyleLedda, Marco, Alessandro Pluchino, and Marco Ragusa. 2024. "Exploring the Role of Genetic and Environmental Features in Colorectal Cancer Development: An Agent-Based Approach" Entropy 26, no. 11: 923. https://doi.org/10.3390/e26110923
APA StyleLedda, M., Pluchino, A., & Ragusa, M. (2024). Exploring the Role of Genetic and Environmental Features in Colorectal Cancer Development: An Agent-Based Approach. Entropy, 26(11), 923. https://doi.org/10.3390/e26110923