
Chemoprotective Effects of Xanthohumol against the Carcinogenic Mycotoxin Aflatoxin B1

 ,
, 
Abstract
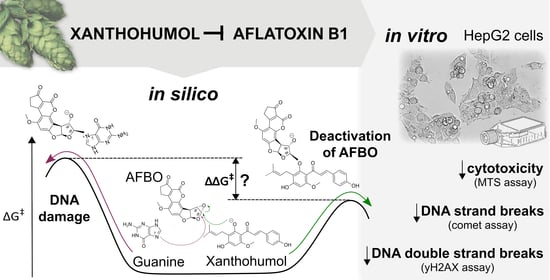
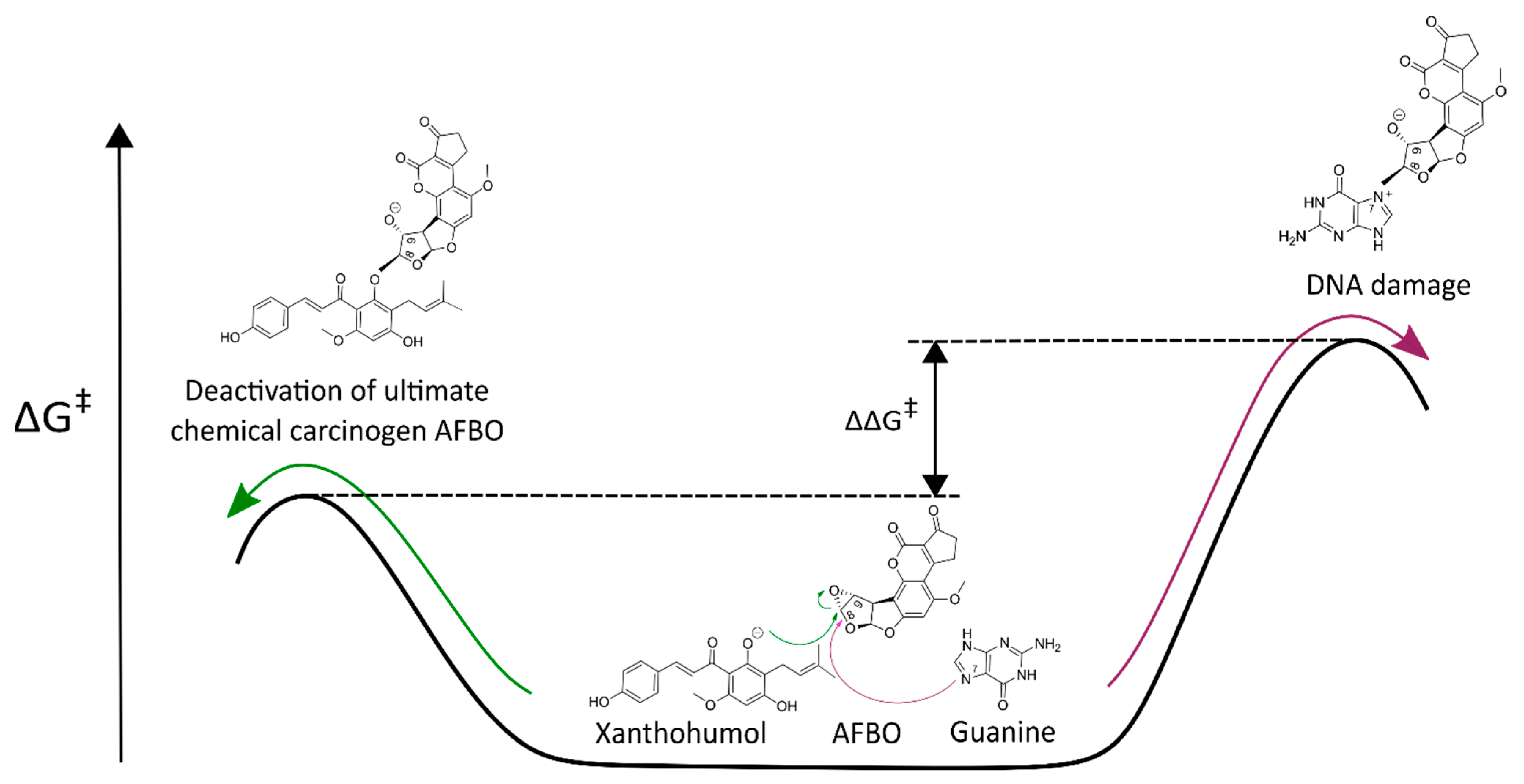
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
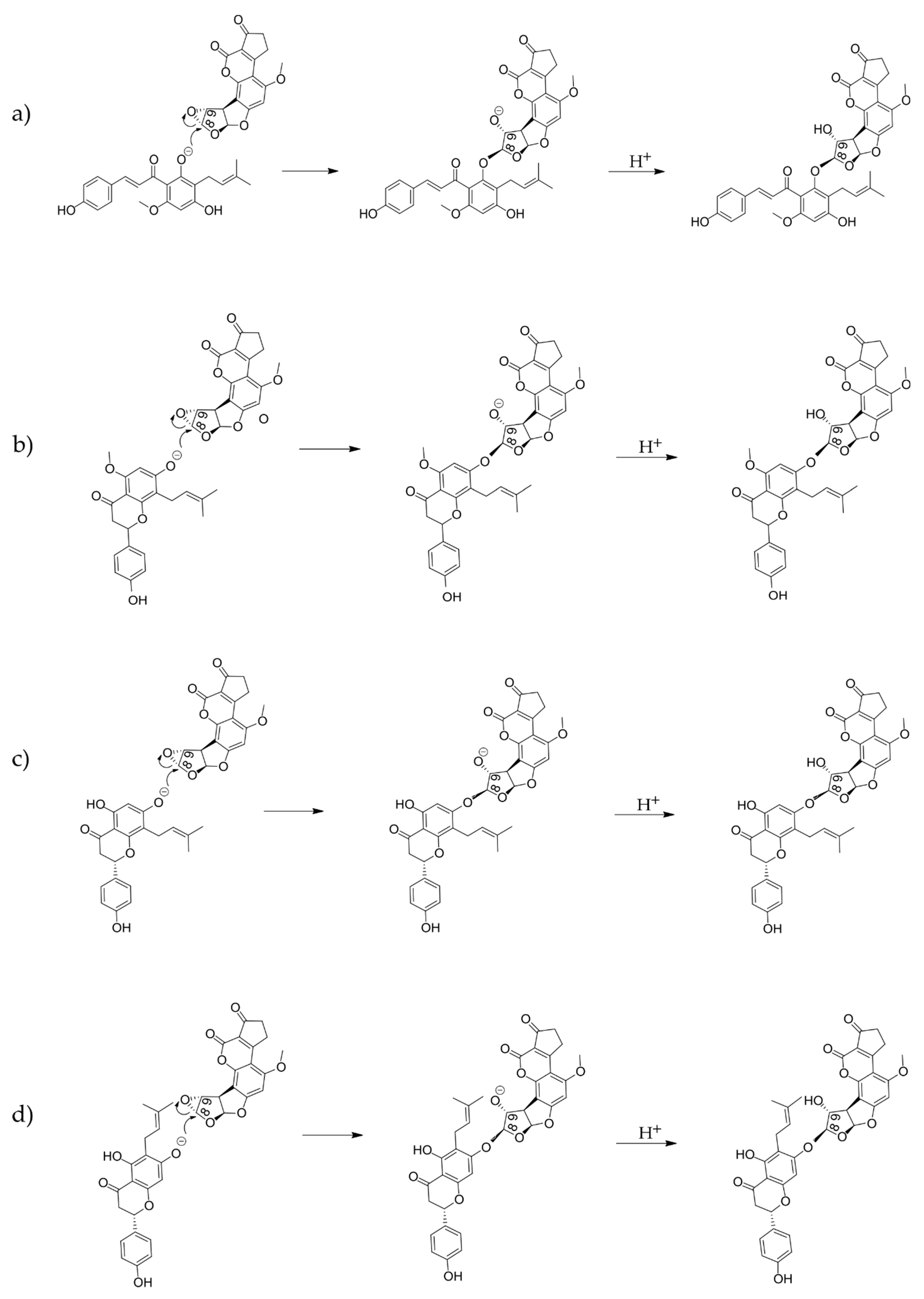
2.2. Computational Analysis of Alkylation Reactions between XN, IXN, 8-PN, and 6-PN and the Carcinogenic Metabolite of AFB1-AFBO
2.3. Cell Culture for the In Vitro Assessment of XN Protective Effects against AFB1
2.4. Changes in Cell Viability—The MTS Assay
2.5. Induction of DNA Strand Breaks—The Alkaline Comet Assay
2.6. Induction of Double Strand Breaks—The γH2AX Assay
2.7. Changes in the Cell Cycle—Analysis by Flow Cytometry
3. Results and Discussion
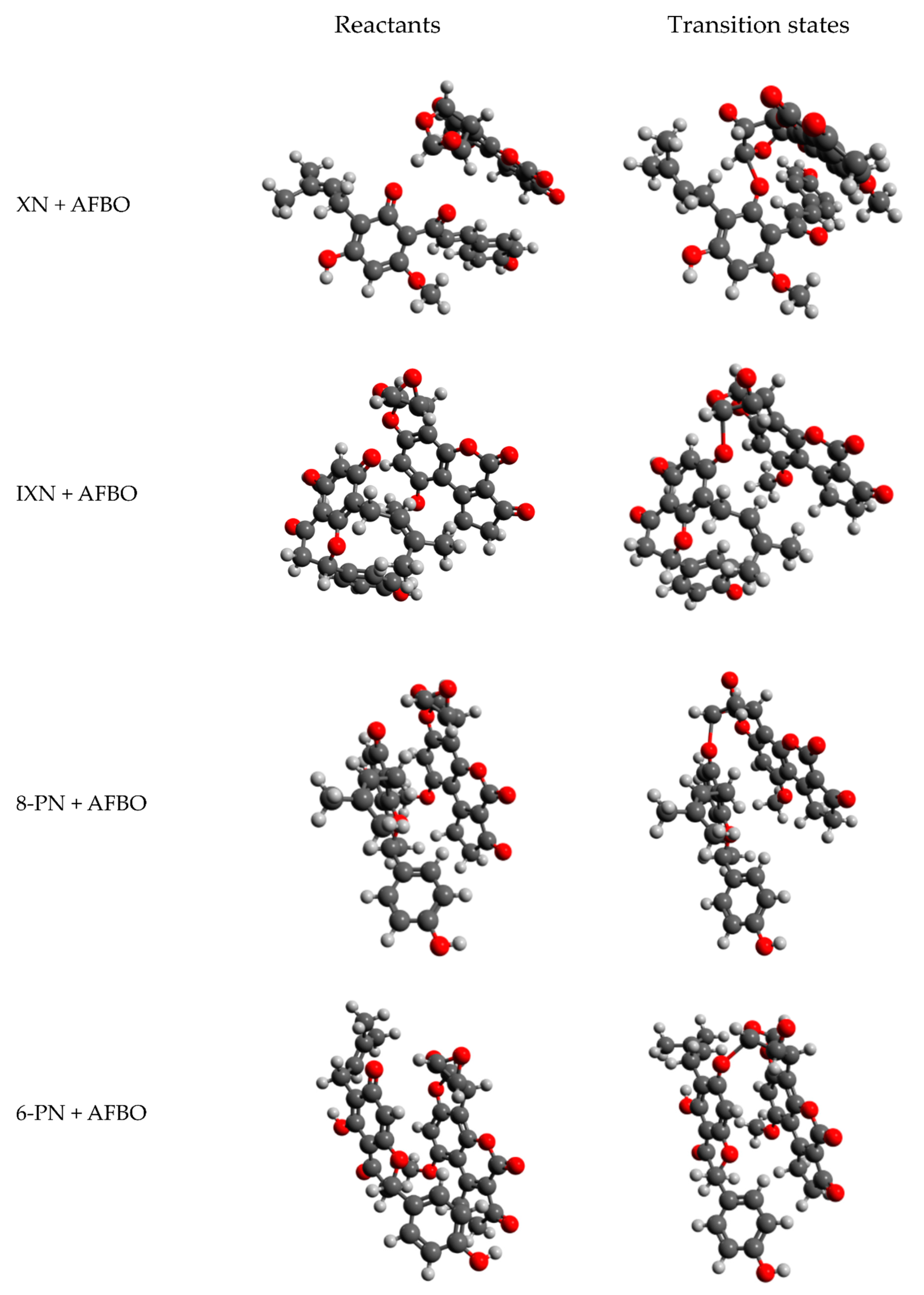
3.1. Mechanistic Insights into Alkylation Reactions of XN, IXN, 8-PN, and 6-PN with the Carcinogenic Metabolite of AFB1-AFBO
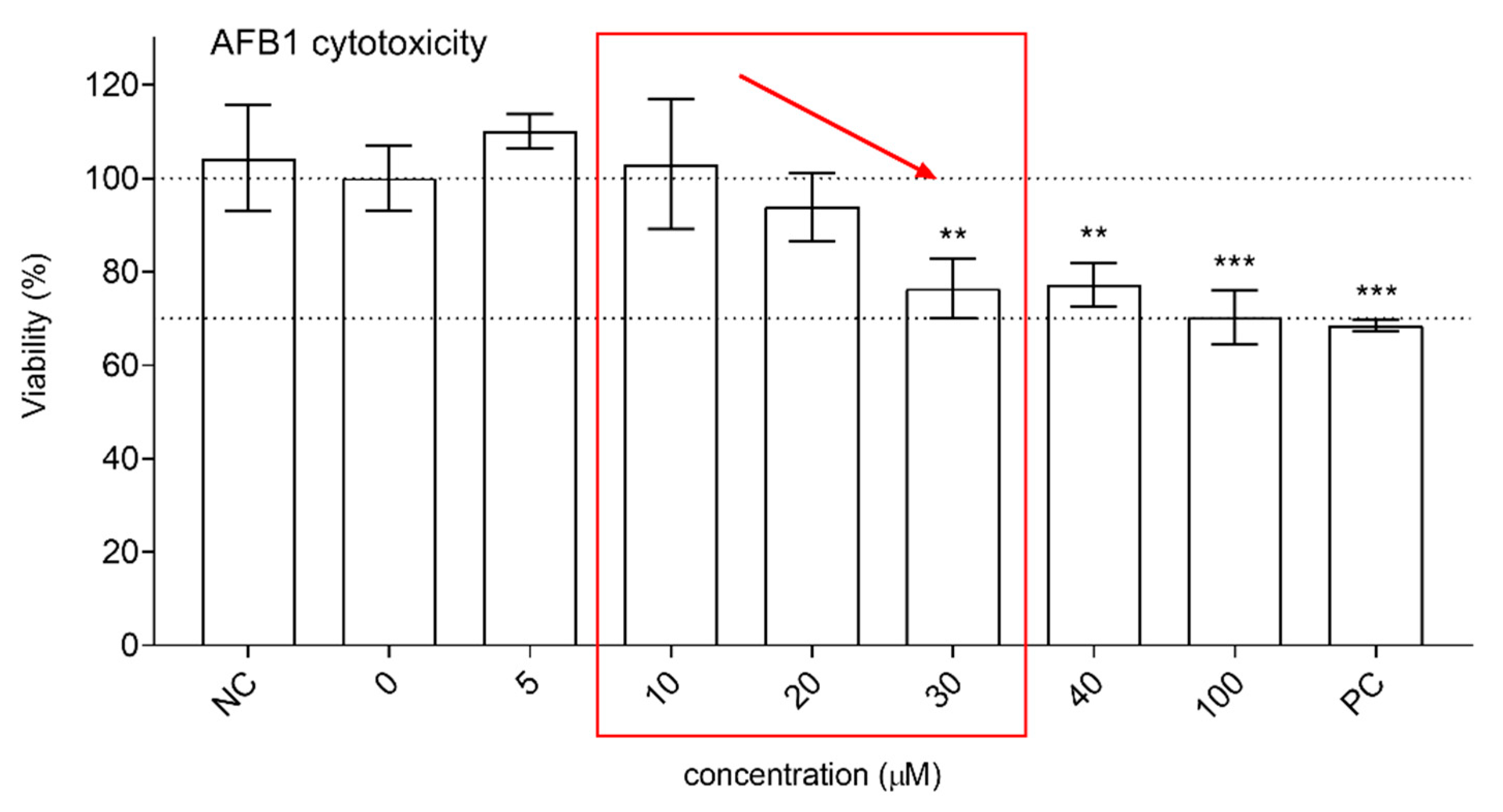
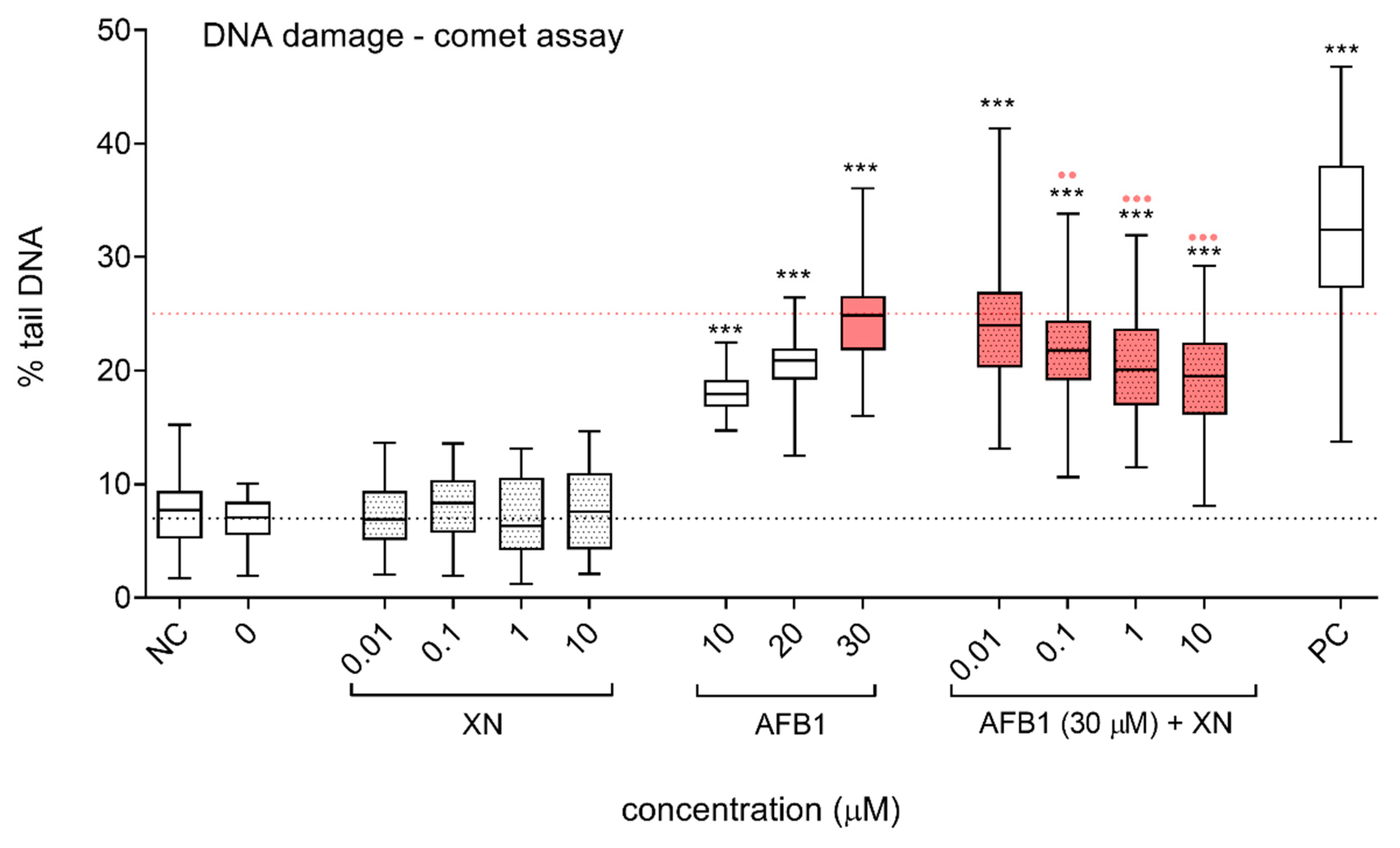
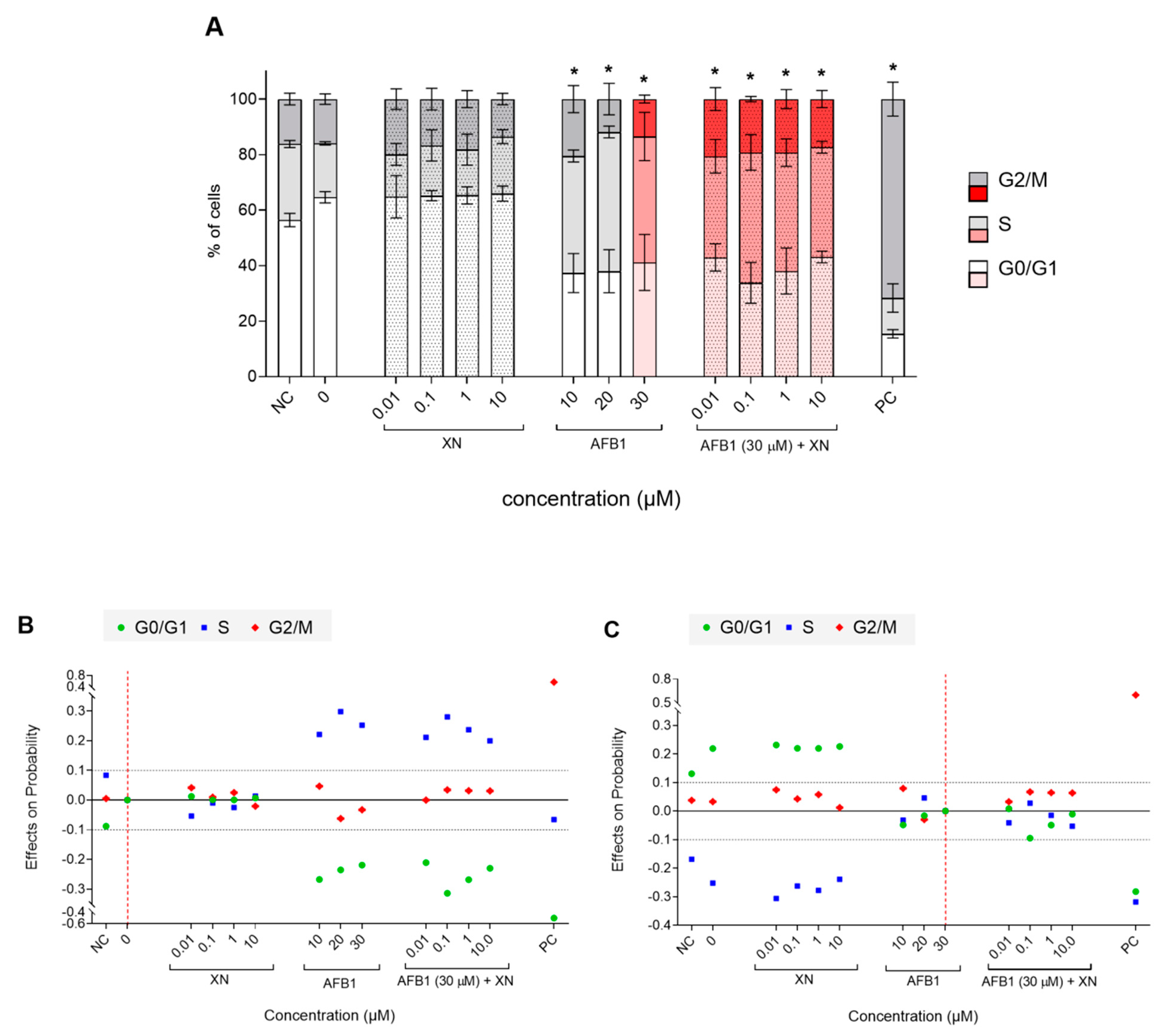
3.2. Protective Effects of XN against AFB1-Induced Cytotoxicity and Genotoxicity In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Hemeryck, L.Y.; Vanhaecke, L. Diet-related DNA adduct formation in relation to carcinogenesis. Nutr. Rev. 2016, 74, 475–489. [Google Scholar] [CrossRef]
- Pezzuto, J.M. Natural Products for Cancer Chemoprevention; Springer Nature Switzerland AG.: Cham, Switzerland, 2020; ISBN 9783030398545. [Google Scholar]
- González-Vallinas, M.; González-Castejón, M.; Rodríguez-Casado, A.; De Molina, A.R. Dietary phytochemicals in cancer prevention and therapy: A complementary approach with promising perspectives. Nutr. Rev. 2013, 71, 585–599. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Yim, D.G.; Lee, D.Y.; Kim, O.Y.; Kang, H.J.; Kim, H.S.; Jang, A.; Park, T.S.; Jin, S.K.; Hur, S.J. Overview of the effect of natural products on reduction of potential carcinogenic substances in meat products. Trends Food Sci. Technol. 2020, 99, 568–579. [Google Scholar] [CrossRef]
- Ramos, S. Cancer chemoprevention and chemotherapy: Dietary polyphenols and signalling pathways. Mol. Nutr. Food Res. 2008, 52, 507–526. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Zhou, T.; Jiang, Y.; Chang, S.K.; Yang, B. Prenylated flavonoids in foods and their applications on cancer prevention. Crit. Rev. Food Sci. Nutr. 2021, 62, 1–14. [Google Scholar] [CrossRef]
- Stevens, J.F. Xanthohumol and Structurally Related Prenylflavonoids for Cancer Chemoprevention and Control. In Natural Products for Cancer Chemoprevention; Springer: Berlin/Heidelberg, Germany, 2020; pp. 319–350. [Google Scholar]
- Boronat, A.; Soldevila-Domenech, N.; Rodríguez-Morató, J.; Martínez-Huélamo, M.; Lamuela-Raventós, R.M.; De La Torre, R. Beer Phenolic Composition of Simple Phenols, Prenylated Flavonoids and Alkylresorcinols. Molecules 2020, 25, 2582. [Google Scholar] [CrossRef]
- Legette, L.; Ma, L.; Reed, R.L.; Miranda, C.L.; Christensen, J.M.; Rodriguez-Proteau, R.; Stevens, J.F. Pharmacokinetics of xanthohumol and metabolites in rats after oral and intravenous administration. Mol. Nutr. Food Res. 2012, 56, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Habschied, K.; Živković, A.; Krstanović, V.; Mastanjević, K. Functional Beer—A Review on Possibilities. Beverages 2020, 6, 51. [Google Scholar] [CrossRef]
- Pinto, C.; Duque, A.L.; Rodríguez-Galdón, B.; Cestero, J.J.; Macías, P. Xanthohumol prevents carbon tetrachloride-induced acute liver injury in rats. Food Chem. Toxicol. 2012, 50, 3405–3412. [Google Scholar] [CrossRef]
- Bolton, J.L.; Dunlap, T.L.; Hajirahimkhan, A.; Mbachu, O.C.; Chen, S.-N.; Chadwick, L.; Nikolic, D.; Van Breemen, R.B.; Pauli, G.F.; Dietz, B.M. The Multiple Biological Targets of Hops and Bioactive Compounds. Chem. Res. Toxicol. 2019, 32, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Ferk, F.; Mišík, M.; Nersesyan, A.; Pichler, C.; Jaeger, W.; Szekeres, T.; Marculescu, R.; Poulsen, H.E.; Henriksen, T.; Bono, R.; et al. Impact of xanthohumol (a prenylated flavonoid from hops) on DNA stability and other health-related biochemical parameters: Results of human intervention trials. Mol. Nutr. Food Res. 2016, 60, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Viegas, O.; Žegura, B.; Pezdric, M.; Novak, M.; Ferreira, I.M.; Pinho, O.; Filipič, M. Protective effects of xanthohumol against the genotoxicity of heterocyclic aromatic amines MeIQx and PhIP in bacteria and in human hepatoma (HepG2) cells. Food Chem. Toxicol. 2012, 50, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Plazar, J.; Žegura, B.; Lah, T.T.; Filipič, M. Protective effects of xanthohumol against the genotoxicity of benzo(a)pyrene (BaP), 2-amino-3-methylimidazo[4,5-f]quinoline (IQ) and tert-butyl hydroperoxide (t-BOOH) in HepG2 human hepatoma cells. Mutat. Res. Toxicol. Environ. Mutagen. 2007, 632, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Plazar, J.; Filipič, M.; Groothuis, G.M. Antigenotoxic effect of Xanthohumol in rat liver slices. Toxicol. Vitr. 2008, 22, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Pichler, C.; Ferk, F.; Al-Serori, H.; Huber, W.; Jager, W.; Waldherr, M.; Mišík, M.; Kundi, M.; Nersesyan, A.; Herbacek, I.; et al. Xanthohumol prevents DNA Damage by dietary carcinogens: Results of a humanintervention trial. Cancer Prev. Res. 2017, 10, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Blot, W.J.; Tarone, R.E. Doll and Peto’s Quantitative Estimates of Cancer Risks: Holding Generally True for 35 Years. J. Natl. Cancer Inst. 2015, 107, 1–5. [Google Scholar] [CrossRef]
- Parkin, D.M. The fraction of cancer attributable to lifestyle and environmental factors in the UK in 2010 Introduction. Br. J. Cancer 2011, 105, 2–5. [Google Scholar] [CrossRef] [Green Version]
- World Cancer Research Fund. American Institute for Cancer Research Diet, Nutrition, Physical Activity and Cancer: A Global Perspective; World Cancer Research Fund International: London, UK, 2018; ISBN 9781912259465. [Google Scholar]
- Wogan, G.N.; Hecht, S.S.; Felton, J.S.; Conney, A.H.; Loeb, L.A. Environmental and chemical carcinogenesis. Semin. Cancer Biol. 2004, 14, 473–486. [Google Scholar] [CrossRef]
- IARC. Aflatoxins IARC Monographs. In IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans; IARC: Lyon, France, 2012; Volume 100, pp. 225–248. ISBN 9789283213239. [Google Scholar]
- IARC. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Amines and Mycotoxins. In IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans; IARC: Lyon, France, 1993; Volume 56, pp. 245–395. [Google Scholar]
- Dai, Y.; Huang, K.; Zhang, B.; Zhu, L.; Xu, W. Aflatoxin B1-induced epigenetic alterations: An overview. Food Chem. Toxicol. 2017, 109, 683–689. [Google Scholar] [CrossRef]
- IARC. Some Traditional Herbal Medicines, Some Mycotoxins, Naphthalene and Styrene. In IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans; IARC: Lyon, France, 2002; Volume 82, pp. 171–301. [Google Scholar]
- Rushing, B.R.; Selim, M.I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem. Toxicol. 2019, 124, 81–100. [Google Scholar] [CrossRef]
- Wang, J.S.; Huang, T.; Su, J.; Liang, F.; Wei, Z.; Liang, Y.; Luo, H.; Kuang, S.Y.; Qian, G.S.; Sun, G.; et al. Hepatocellular carcinoma and aflatoxin exposure in Zhuqing Village, Fusui County, People’s Republic of China. Cancer Epidemiol. Biomark. Prev. 2001, 10, 143–146. [Google Scholar]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [Green Version]
- Kew, M.C. Aflatoxins as a Cause of Hepatocellular Carcinoma. J. Gastrointest. Liver Dis. 2013, 22, 305–310. [Google Scholar]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Florian, J.; Warshel, A. Langevin Dipoles Model for ab Initio Calculations of Chemical Processes in Solution: Parametrization and Application to Hydration Free Energies of Neutral and Ionic Solutes and Conformational Analysis in Aqueous Solution. J. Phys. Chem. B 1997, 101, 5583–5595. [Google Scholar] [CrossRef]
- Borštnik, U.; Janežič, D. Symplectic Molecular Dynamics Simulations on Specially Designed Parallel Computers. J. Chem. Inf. Model. 2005, 45, 1600–1604. [Google Scholar] [CrossRef]
- Borštnik, U.; Hodošček, M.; Janežič, D. Improving the performance of molecular dynamics simulations on parallel clusters. J. Chem. Inf. Comput. Sci. 2004, 44, 359–364. [Google Scholar] [CrossRef]
- Bren, U.; Guengerich, F.P.; Mavri, J. Guanine Alkylation by the Potent Carcinogen Aflatoxin B1: Quantum Chemical Calculations. Chem. Res. Toxicol. 2007, 20, 1134–1140. [Google Scholar] [CrossRef]
- Lajovic, A.; Nagy, L.D.; Guengerich, F.P.; Bren, U. Carcinogenesis of Urethane: Simulation versus Experiment. Chem. Res. Toxicol. 2015, 28, 691–701. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 16, revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Furlan, V.; Bren, U. Protective Effects of [6]-Gingerol Against Chemical Carcinogens: Mechanistic Insights. Int. J. Mol. Sci. 2020, 21, 695. [Google Scholar] [CrossRef] [Green Version]
- MarvinSketch. Version 18.5.0, Developed by ChemAxon. 2019. Available online: http://www.chemaxon.com/products/marvin/marvinsketch/ (accessed on 1 May 2021).
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hercog, K.; Maisanaba, S.; Filipič, M.; Sollner-Dolenc, M.; Kač, L.; Žegura, B. Genotoxic activity of bisphenol A and its analogues bisphenol S, bisphenol F and bisphenol AF and their mixtures in human hepatocellular carcinoma (HepG2) cells. Sci. Total Environ. 2019, 687, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Žegura, B.; Filipič, M. Application of In Vitro Comet Assay for Genotoxicity Testing. In Optimization in Drug Discovery; Humana Press: Totowa, NJ, USA, 2004; pp. 301–313. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. Reshaping Data with thereshapePackage. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D. R Development Core Team. 2010. nlme: Linear and Nonlinear Mixed Effects Models. R Packag. Version 2007, 3, 1–89. [Google Scholar]
- Guo, J.; Nikolić, D.; Chadwick, L.R.; Pauli, G.F.; Van Breemen, R.B. Identification of Human Hepatic Cytochrome p450 Enzymes Involved in the Metabolism of 8-Prenylnaringenin and Isoxanthohumol from Hops (Humulus Lupulus L.). Drug Metab. Dispos. 2006, 34, 1152–1159. [Google Scholar] [CrossRef] [Green Version]
- Bren, U.; Zupan, M.; Guengerich, F.P.; Mavri, J. Chemical Reactivity as a Tool to Study Carcinogenicity: Reaction between Chloroethylene Oxide and Guanine. J. Org. Chem. 2006, 71, 4078–4084. [Google Scholar] [CrossRef]
- Galeša, K.; Bren, U.; Kranjc, A.; Mavri, J. Carcinogenicity of Acrylamide: A Computational Study. J. Agric. Food Chem. 2008, 56, 8720–8727. [Google Scholar] [CrossRef]
- McCullough, A.K.; Lloyd, R.S. Mechanisms underlying aflatoxin-associated mutagenesis—Implications in carcinogenesis. DNA Repair 2019, 77, 76–86. [Google Scholar] [CrossRef]
- Waldherr, M.; Mišík, M.; Ferk, F.; Tomc, J.; Žegura, B.; Filipič, M.; Mikulits, W.; Mai, S.; Haas, O.; Huber, W.W.; et al. Use of HuH6 and other human-derived hepatoma lines for the detection of genotoxins: A new hope for laboratory animals? Arch. Toxicol. 2018, 92, 921–934. [Google Scholar] [CrossRef] [Green Version]
- Westerink, W.M.; Schoonen, W.G. Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol. Vitr. 2007, 21, 1581–1591. [Google Scholar] [CrossRef]
- Žegura, B.; Heath, E.; Černoša, A.; Filipič, M. Combination of in vitro bioassays for the determination of cytotoxic and genotoxic potential of wastewater, surface water and drinking water samples. Chemosphere 2009, 75, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Ferk, F.; Huber, W.W.; Filipič, M.; Bichler, J.; Haslinger, E.; Mišík, M.; Nersesyan, A.; Grasl-Kraupp, B.; Žegura, B.; Knasmüller, S. Xanthohumol, a prenylated flavonoid contained in beer, prevents the induction of preneoplastic lesions and DNA damage in liver and colon induced by the heterocyclic aromatic amine amino-3-methyl-imidazo[4,5-f]quinoline (IQ). Mutat. Res. Mol. Mech. Mutagen. 2010, 691, 17–22. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Kopp, B.; Khoury, L.; Audebert, M. Validation of the γH2AX biomarker for genotoxicity assessment: A review. Arch. Toxicol. 2019, 93, 2103–2114. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA Double-stranded Breaks Induce Histone H2AX Phosphorylation on Serine. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase Chromatin Domains Involved in DNA Double-Strand Breaks in Vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, Z.; Wang, X.; Wang, Y.; Zhang, X.; Lu, H.; Wang, S.-L. Cytochrome P450 2A13 enhances the sensitivity of human bronchial epithelial cells to aflatoxin B1-induced DNA damage. Toxicol. Appl. Pharmacol. 2013, 270, 114–121. [Google Scholar] [CrossRef]
- Huang, B.; Chen, Q.; Wang, L.; Gao, X.; Zhu, W.; Mu, P.; Deng, Y. Aflatoxin B1 Induces Neurotoxicity through Reactive Oxygen Species Generation, DNA Damage, Apoptosis, and S-Phase Cell Cycle Arrest. Int. J. Mol. Sci. 2020, 21, 6517. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Gao, J.; Huang, K.; Luo, Y.; Zhang, B.; Xu, W. miR-34a screened by miRNA profiling negatively regulates Wnt/β-catenin signaling pathway in Aflatoxin B1 induced hepatotoxicity. Sci. Rep. 2015, 5, 16732. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Lv, Y.; Huang, K.; Luo, Y.; Xu, W. Zinc inhibits aflatoxin B1-induced cytotoxicity and genotoxicity in human hepatocytes (HepG2 cells). Food Chem. Toxicol. 2016, 92, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef]
- Gerhauser, C.; Alt, A.; Heiss, E.; Gamal-Eldeen, A.; Klimo, K.; Knauft, J.; Neumann, I.; Scherf, H.-R.; Frank, N.; Bartsch, H.; et al. Cancer chemopreventive activity of Xanthohumol, a natural product derived from hop. Mol. Cancer Ther. 2002, 1, 959–969. [Google Scholar]
- Henderson, M.C.; Miranda, C.L.; Stevens, J.F.; Deinzer, M.L.; Buhler, D.R. In Vitro Inhibition of Human P450 Enzymes by Prenylated Flavonoids from Hops, Humulus Lupulus. Xenobiotica 2000, 30, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Gross-Steinmeyer, K.; Stapleton, P.L.; Tracy, J.H.; Bammler, T.K.; Strom, S.; Buhler, D.R.; Eaton, D.L. Modulation of Aflatoxin B1–Mediated Genotoxicity in Primary Cultures of Human Hepatocytes by Diindolylmethane, Curcumin and Xanthohumols. Toxicol. Sci. 2009, 112, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyphenolic Scavenger | Aflatoxin B1 exo-8,9-epoxide | ||
|---|---|---|---|
| HF/6-311++G(d,p) | [kcal/mol] | [kcal/mol] | [kcal/mol] |
| XN | 21.06 | 14.70 | |
| IXN | 21.72 | 14.44 | |
| 8-PN | 24.56 | 14.88 | |
| 6-PN | 23.73 | 13.14 | |
| Guanine | 28.90 | 14.25 | 15.1 [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štern, A.; Furlan, V.; Novak, M.; Štampar, M.; Kolenc, Z.; Kores, K.; Filipič, M.; Bren, U.; Žegura, B. Chemoprotective Effects of Xanthohumol against the Carcinogenic Mycotoxin Aflatoxin B1. Foods 2021, 10, 1331. https://doi.org/10.3390/foods10061331
Štern A, Furlan V, Novak M, Štampar M, Kolenc Z, Kores K, Filipič M, Bren U, Žegura B. Chemoprotective Effects of Xanthohumol against the Carcinogenic Mycotoxin Aflatoxin B1. Foods. 2021; 10(6):1331. https://doi.org/10.3390/foods10061331
Chicago/Turabian StyleŠtern, Alja, Veronika Furlan, Matjaž Novak, Martina Štampar, Zala Kolenc, Katarina Kores, Metka Filipič, Urban Bren, and Bojana Žegura. 2021. "Chemoprotective Effects of Xanthohumol against the Carcinogenic Mycotoxin Aflatoxin B1" Foods 10, no. 6: 1331. https://doi.org/10.3390/foods10061331