
The Evolving Roles of Cardiac Macrophages in Homeostasis, Regeneration, and Repair


Abstract
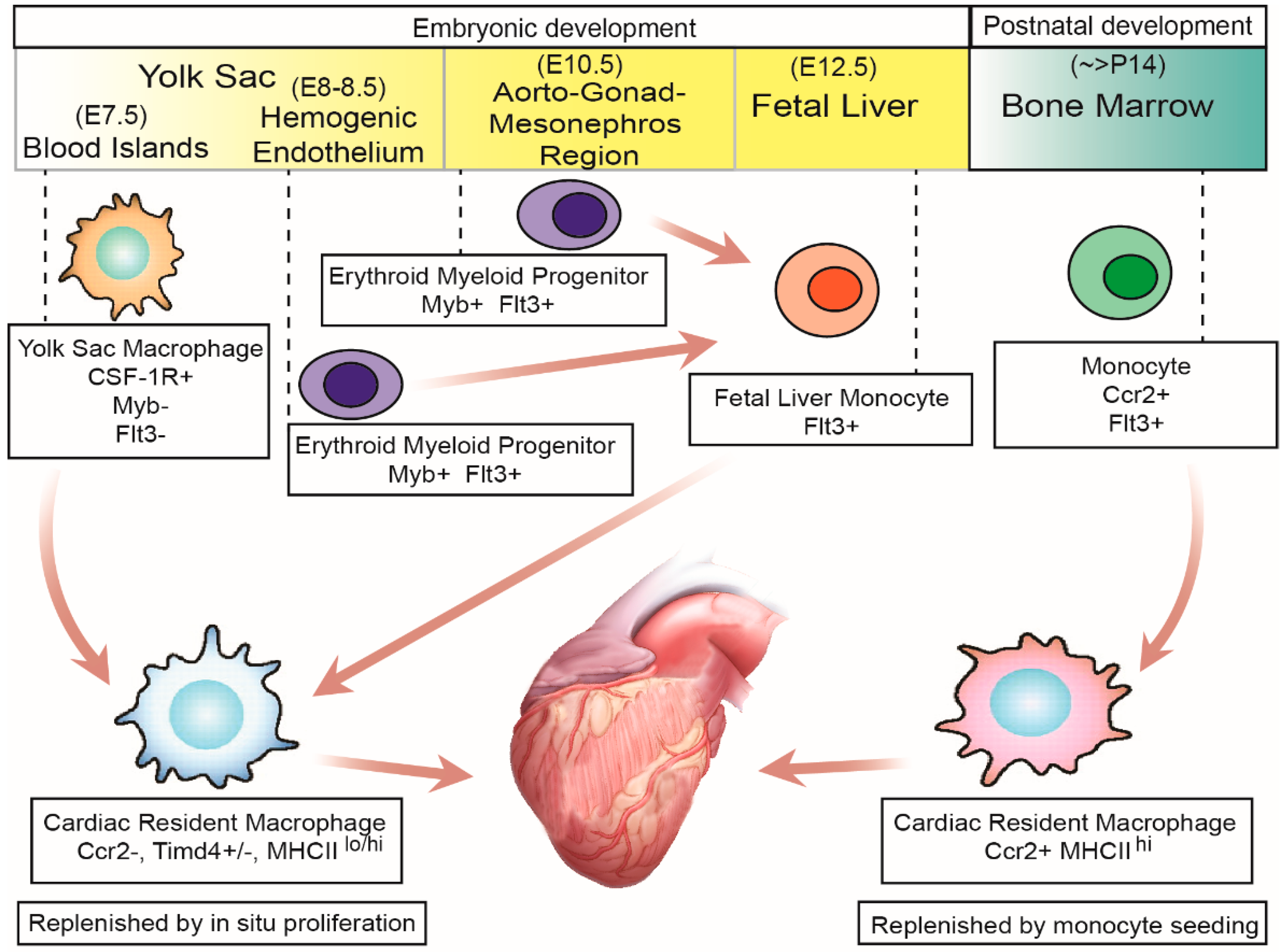
:1. Introduction
2. Origin and Development of Cardiac Resident Macrophages
3. Advancements in Cardiac Resident Macrophage Characterization
4. Homeostatic Impact of Cardiac Macrophages
4.1. Capillary Development and Lymphatic Network Maturation
4.2. Electrical Conduction in the Heart
4.3. Mitochondrial Function
5. Role of Cardiac Macrophages Following Adult Cardiac Injury
5.1. Ischemic Heart Failure
5.2. Angiotensin Infusion and Pressure Overload
5.3. Graft Rejection Following Heart Transplant
5.4. Chemotherapeutic Cardiotoxicity
6. Cardiac Macrophages in Cardiac Regeneration
7. Future Perspectives
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tauber, A.I. Metchnikoff and the phagocytosis theory. Nat. Rev. Mol. Cell Biol. 2003, 4, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar]
- Davies, L.C.; Taylor, P.R. Tissue-resident macrophages: Then and now. Immunology 2015, 144, 541–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J. Exp. Med. 2012, 209, 1167–1181. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Gong, Y.; Huang, T.; Lee, C.Z.W.; Bian, L.; Bai, Z.; Shi, H.; Zeng, Y.; Liu, C.; He, J.; et al. Deciphering human macrophage development at single-cell resolution. Nature 2020, 582, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, C.; Ginhoux, F. Understanding the Heterogeneity of Resident Liver Macrophages. Front. Immunol. 2019, 10, 2694. [Google Scholar] [CrossRef] [Green Version]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- De Schepper, S.; Verheijden, S.; Aguilera-Lizarraga, J.; Viola, M.F.; Boesmans, W.; Stakenborg, N.; Voytyuk, I.; Schmidt, I.; Boeckx, B.; Dierckx de Casterle, I.; et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell 2018, 175, 400–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mege, J.L.; Mehraj, V.; Capo, C. Macrophage polarization and bacterial infections. Curr. Opin. Infect. Dis. 2011, 24, 230–234. [Google Scholar] [CrossRef]
- Geissmann, F.; Gordon, S.; Hume, D.A.; Mowat, A.M.; Randolph, G.J. Unravelling mononuclear phagocyte heterogeneity. Nat. Rev. Immunol. 2010, 10, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Sreejit, G.; Fleetwood, A.J.; Murphy, A.J.; Nagareddy, P.R. Origins and diversity of macrophages in health and disease. Clin. Transl. Immunol. 2020, 9, e1222. [Google Scholar] [CrossRef]
- Stuart, L.M.; Ezekowitz, R.A. Phagocytosis: Elegant complexity. Immunity 2005, 22, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. Biomed. Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Pluddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Pinto, A.R.; Epelman, S.; Kopecky, B.J.; Clemente-Casares, X.; Godwin, J.; Rosenthal, N.; Kovacic, J.C. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 2018, 72, 2213–2230. [Google Scholar] [CrossRef]
- Pinto, A.R.; Paolicelli, R.; Salimova, E.; Gospocic, J.; Slonimsky, E.; Bilbao-Cortes, D.; Godwin, J.W.; Rosenthal, N.A. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 2012, 7, e36814. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; van der Laan, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austyn, J.M.; Gordon, S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 1981, 11, 805–815. [Google Scholar] [CrossRef]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.M.; Ryan, G.R.; Hapel, A.J.; Dominguez, M.G.; Russell, R.G.; Kapp, S.; Sylvestre, V.; Stanley, E.R. Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood 2002, 99, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Pridans, C.; Raper, A.; Davis, G.M.; Alves, J.; Sauter, K.A.; Lefevre, L.; Regan, T.; Meek, S.; Sutherland, L.; Thomson, A.J.; et al. Pleiotropic Impacts of Macrophage and Microglial Deficiency on Development in Rats with Targeted Mutation of the Csf1r Locus. J. Immunol. 2018, 201, 2683–2699. [Google Scholar] [CrossRef] [Green Version]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Rojo, R.; Raper, A.; Ozdemir, D.D.; Lefevre, L.; Grabert, K.; Wollscheid-Lengeling, E.; Bradford, B.; Caruso, M.; Gazova, I.; Sanchez, A.; et al. Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nat. Commun. 2019, 10, 3215. [Google Scholar] [CrossRef]
- Kumaravelu, P.; Hook, L.; Morrison, A.M.; Ure, J.; Zhao, S.; Zuyev, S.; Ansell, J.; Medvinsky, A. Quantitative developmental anatomy of definitive haematopoietic stem cells/long-term repopulating units (HSC/RUs): Role of the aorta-gonad-mesonephros (AGM) region and the yolk sac in colonisation of the mouse embryonic liver. Development 2002, 129, 4891–4899. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, L.; Saelens, W.; De Prijck, S.; Martens, L.; Scott, C.L.; Van Isterdael, G.; Hoffmann, E.; Beyaert, R.; Saeys, Y.; Lambrecht, B.N.; et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 2016, 44, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine. Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef]
- Ajuebor, M.N.; Flower, R.J.; Hannon, R.; Christie, M.; Bowers, K.; Verity, A.; Perretti, M. Endogenous monocyte chemoattractant protein-1 recruits monocytes in the zymosan peritonitis model. J. Leukoc. Biol. 1998, 63, 108–116. [Google Scholar] [CrossRef]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef]
- Dawson, T.C.; Kuziel, W.A.; Osahar, T.A.; Maeda, N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 1999, 143, 205–211. [Google Scholar] [CrossRef]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Xu, A.; Sun, X.; Yang, Y.; Zhang, L.; Bai, H.; Ben, J.; Zhu, X.; Li, X.; Yang, Q.; et al. Self-Maintenance of Cardiac Resident Reparative Macrophages Attenuates Doxorubicin-Induced Cardiomyopathy Through the SR-A1-c-Myc Axis. Circ. Res. 2020, 127, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Zaman, R.; Hamidzada, H.; Epelman, S. Exploring cardiac macrophage heterogeneity in the healthy and diseased myocardium. Curr. Opin. Immunol. 2020, 68, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef] [Green Version]
- Discher, D.E.; Janmey, P.; Wang, Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, H.; Aikawa, M.; Hill, C.C.; Weiss, D.; Taylor, W.R.; Libby, P.; Lee, R.T. Biomechanical strain induces class a scavenger receptor expression in human monocyte/macrophages and THP-1 cells: A potential mechanism of increased atherosclerosis in hypertension. Circulation 2001, 104, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fereol, S.; Fodil, R.; Labat, B.; Galiacy, S.; Laurent, V.M.; Louis, B.; Isabey, D.; Planus, E. Sensitivity of alveolar macrophages to substrate mechanical and adhesive properties. Cell Motil. Cytoskelet. 2006, 63, 321–340. [Google Scholar] [CrossRef]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.; Lavine, K.J. Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2016, 118, 368–370. [Google Scholar] [CrossRef]
- Nicolas-Avila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martinez, L.; Sanchez-Diaz, M.; Diaz-Garcia, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wulfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.H.; Koh, Y.J.; Han, J.; Sung, H.K.; Jong Lee, H.; Morisada, T.; Schwendener, R.A.; Brekken, R.A.; Kang, G.; Oike, Y.; et al. Angiogenic role of LYVE-1-positive macrophages in adipose tissue. Circ. Res. 2007, 100, e47–e57. [Google Scholar] [CrossRef] [Green Version]
- Gula, G.; Ruminski, S.; Niderla-Bielinska, J.; Jasinska, A.; Kiernozek, E.; Jankowska-Steifer, E.; Flaht-Zabost, A.; Ratajska, A. Potential functions of embryonic cardiac macrophages in angiogenesis, lymphangiogenesis and extracellular matrix remodeling. Histochem. Cell Biol. 2020, 1155, 117–132. [Google Scholar] [CrossRef]
- Cahill, T.J.; Sun, X.; Ravaud, C.; Villa Del Campo, C.; Klaourakis, K.; Lupu, I.E.; Lord, A.M.; Browne, C.; Jacobsen, S.E.W.; Greaves, D.R.; et al. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development 2021, 148, dev194563. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.; Crocker, P.R.; Gordon, S. Murine fetal liver macrophages bind developing erythroblasts by a divalent cation-dependent hemagglutinin. J. Cell Biol. 1988, 106, 649–656. [Google Scholar] [CrossRef] [Green Version]
- De Back, D.Z.; Kostova, E.B.; van Kraaij, M.; van den Berg, T.K.; van Bruggen, R. Of macrophages and red blood cells; a complex love story. Front. Physiol. 2014, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Koscso, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.M.; Mucida, D.; et al. Crosstalk between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell 2014, 158, 1210. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Macrophages and Iron Metabolism. Microbiol. Spectr. 2016, 4, 492–504. [Google Scholar] [CrossRef]
- Squarzoni, P.; Oller, G.; Hoeffel, G.; Pont-Lezica, L.; Rostaing, P.; Low, D.; Bessis, A.; Ginhoux, F.; Garel, S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014, 8, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Dissing-Olesen, L.; Stevens, B. New insights on the role of microglia in synaptic pruning in health and disease. Curr. Opin. Neurobiol. 2016, 36, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Polavarapu, R.; Eskla, K.L.; Pantner, Y.; Nicholson, C.K.; Ishii, M.; Brunnhoelzl, D.; Mauria, R.; Husain, A.; Naqvi, N.; et al. Impact of Lymphangiogenesis on Cardiac Remodeling After Ischemia and Reperfusion Injury. J. Am. Heart Assoc. 2018, 7, e009565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houssari, M.; Dumesnil, A.; Tardif, V.; Kivela, R.; Pizzinat, N.; Boukhalfa, I.; Godefroy, D.; Schapman, D.; Hemanthakumar, K.A.; Bizou, M.; et al. Lymphatic and Immune Cell Cross-Talk Regulates Cardiac Recovery After Experimental Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1722–1737. [Google Scholar] [CrossRef]
- Harrison, M.R.; Feng, X.; Mo, G.; Aguayo, A.; Villafuerte, J.; Yoshida, T.; Pearson, C.A.; Schulte-Merker, S.; Lien, C.L. Late developing cardiac lymphatic vasculature supports adult zebrafish heart function and regeneration. eLife 2019, 8, e42762. [Google Scholar] [CrossRef]
- Sugita, J.; Fujiu, K.; Nakayama, Y.; Matsubara, T.; Matsuda, J.; Oshima, T.; Liu, Y.; Maru, Y.; Hasumi, E.; Kojima, T.; et al. Cardiac macrophages prevent sudden death during heart stress. Nat. Commun. 2021, 12, 1910. [Google Scholar] [CrossRef]
- Chen, B.; Frangogiannis, N.G. Immune cells in repair of the infarcted myocardium. Microcirculation 2017, 24, e12305. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cell biological mechanisms in regulation of the post-infarction inflammatory response. Curr. Opin. Physiol. 2018, 1, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M. Myeloid cells in cardiovascular organs. J. Intern. Med. 2019, 285, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef]
- De Couto, G. Macrophages in cardiac repair: Environmental cues and therapeutic strategies. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kyne, L.; Hausdorff, J.M.; Knight, E.; Dukas, L.; Azhar, G.; Wei, J.Y. Neutrophilia and congestive heart failure after acute myocardial infarction. Am. Heart J. 2000, 139, 94–100. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Frodermann, V.; Nahrendorf, M. Neutrophil-macrophage cross-talk in acute myocardial infarction. Eur. Heart J. 2017, 38, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilgendorf, I.; Gerhardt, L.M.; Tan, T.C.; Winter, C.; Holderried, T.A.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Courties, G.; Heidt, T.; Sebas, M.; Iwamoto, Y.; Jeon, D.; Truelove, J.; Tricot, B.; Wojtkiewicz, G.; Dutta, P.; Sager, H.B.; et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J. Am. Coll. Cardiol. 2014, 63, 1556–1566. [Google Scholar] [CrossRef]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef] [Green Version]
- Daseke, M.J., II; Tenkorang-Impraim, M.A.A.; Ma, Y.; Chalise, U.; Konfrst, S.R.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Exogenous IL-4 shuts off pro-inflammation in neutrophils while stimulating anti-inflammation in macrophages to induce neutrophil phagocytosis following myocardial infarction. J. Mol. Cell Cardiol. 2020, 145, 112–121. [Google Scholar] [CrossRef]
- Ferraro, B.; Leoni, G.; Hinkel, R.; Ormanns, S.; Paulin, N.; Ortega-Gomez, A.; Viola, J.R.; de Jong, R.; Bongiovanni, D.; Bozoglu, T.; et al. Pro-Angiogenic Macrophage Phenotype to Promote Myocardial Repair. J. Am. Coll. Cardiol. 2019, 73, 2990–3002. [Google Scholar] [CrossRef]
- DeBerge, M.; Yeap, X.Y.; Dehn, S.; Zhang, S.; Grigoryeva, L.; Misener, S.; Procissi, D.; Zhou, X.; Lee, D.C.; Muller, W.A.; et al. MerTK Cleavage on Resident Cardiac Macrophages Compromises Repair After Myocardial Ischemia Reperfusion Injury. Circ. Res. 2017, 121, 930–940. [Google Scholar] [CrossRef]
- Van der Laan, A.M.; Ter Horst, E.N.; Delewi, R.; Begieneman, M.P.; Krijnen, P.A.; Hirsch, A.; Lavaei, M.; Nahrendorf, M.; Horrevoets, A.J.; Niessen, H.W.; et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur. Heart J. 2014, 35, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuschner, F.; Rauch, P.J.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef]
- Jenca, D.; Melenovsky, V.; Stehlik, J.; Stanek, V.; Kettner, J.; Kautzner, J.; Adamkova, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2020, 24. [Google Scholar] [CrossRef]
- Forte, E.; Panahi, M.; Baxan, N.; Ng, F.S.; Boyle, J.J.; Branca, J.; Bedard, O.; Hasham, M.G.; Benson, L.; Harding, S.E.; et al. Type 2 MI induced by a single high dose of isoproterenol in C57BL/6J mice triggers a persistent adaptive immune response against the heart. J. Cell Mol. Med. 2021, 25, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Tacke, R.; Hedrick, C.C.; Hanna, R.N. Nonclassical patrolling monocyte function in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Buscher, K.; Marcovecchio, P.; Hedrick, C.C.; Ley, K. Patrolling Mechanics of Non-Classical Monocytes in Vascular Inflammation. Front. Cardiovasc. Med. 2017, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Simoes, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 2020, 11, 600. [Google Scholar] [CrossRef] [Green Version]
- Bevan, L.; Lim, Z.W.; Venkatesh, B.; Riley, P.R.; Martin, P.; Richardson, R.J. Specific macrophage populations promote both cardiac scar deposition and subsequent resolution in adult zebrafish. Cardiovasc. Res. 2020, 116, 1357–1371. [Google Scholar] [CrossRef] [Green Version]
- Mellak, S.; Ait-Oufella, H.; Esposito, B.; Loyer, X.; Poirier, M.; Tedder, T.F.; Tedgui, A.; Mallat, Z.; Potteaux, S. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe−/− mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Fujiu, K.; Shibata, M.; Nakayama, Y.; Ogata, F.; Matsumoto, S.; Noshita, K.; Iwami, S.; Nakae, S.; Komuro, I.; Nagai, R.; et al. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat. Med. 2017, 23, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, P.B.; Rayburn, B.K.; Hutchins, G.M.; Kasper, E.K.; Baughman, K.L.; Goodman, S.N.; Lecks, L.E.; Baumgartner, W.A.; Hruban, R.H. Peritransplant injury to the myocardium associated with the development of accelerated arteriosclerosis in heart transplant recipients. Am. J. Surg. Pathol. 1994, 18, 338–346. [Google Scholar] [CrossRef]
- Patel, J.K.; Kittleson, M.; Kobashigawa, J.A. Cardiac allograft rejection. Surgeon 2011, 9, 160–167. [Google Scholar] [CrossRef]
- Thornley, T.B.; Fang, Z.; Balasubramanian, S.; Larocca, R.A.; Gong, W.; Gupta, S.; Csizmadia, E.; Degauque, N.; Kim, B.S.; Koulmanda, M.; et al. Fragile TIM-4-expressing tissue resident macrophages are migratory and immunoregulatory. J. Clin. Investig. 2014, 124, 3443–3454. [Google Scholar] [CrossRef] [Green Version]
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124. [Google Scholar] [CrossRef] [Green Version]
- Bansal, S.S.; Ismahil, M.A.; Goel, M.; Patel, B.; Hamid, T.; Rokosh, G.; Prabhu, S.D. Activated T Lymphocytes are Essential Drivers of Pathological Remodeling in Ischemic Heart Failure. Circ. Heart Fail. 2017, 10, e003688. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Chang, H.M.; Moudgil, R.; Scarabelli, T.; Okwuosa, T.M.; Yeh, E.T.H. Cardiovascular Complications of Cancer Therapy: Best Practices in Diagnosis, Prevention, and Management: Part 1. J. Am. Coll. Cardiol. 2017, 70, 2536–2551. [Google Scholar] [CrossRef]
- Kim, S.Y.; Nair, M.G. Macrophages in wound healing: Activation and plasticity. Immunol. Cell. Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef]
- Allard, B.; Panariti, A.; Martin, J.G. Alveolar Macrophages in the Resolution of Inflammation, Tissue Repair, and Tolerance to Infection. Front. Immunol. 2018, 9, 1777. [Google Scholar] [CrossRef] [Green Version]
- Hine, A.M.; Loke, P. Intestinal Macrophages in Resolving Inflammation. J. Immunol. 2019, 203, 593–599. [Google Scholar] [CrossRef]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.M.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef] [Green Version]
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef]
- Lam, N.T.; Sadek, H.A. Neonatal Heart Regeneration: Comprehensive Literature Review. Circulation 2018, 138, 412–423. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, A.I.; Porrello, E.R.; Kimura, W.; Olson, E.N.; Sadek, H.A. Surgical models for cardiac regeneration in neonatal mice. Nat. Protoc. 2014, 9, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Sadek, H.A.; Martin, J.F.; Takeuchi, J.K.; Leor, J.; Nie, Y.; Giacca, M.; Lee, R.T. Multi-investigator letter on reproducibility of neonatal heart regeneration following apical resection. Stem Cell Rep. 2014, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Hou, J. Apical Resection Mouse Model to Study Early Mammalian Heart Regeneration. J. Vis. Exp. 2016, 107, e53488. [Google Scholar] [CrossRef]
- Bryant, D.M.; O Meara, C.C.; Ho, N.N.; Gannon, J.; Cai, L.; Lee, R.T. A systematic analysis of neonatal mouse heart regeneration after apical resection. J. Mol. Cell. Cardiol. 2015, 79, 315–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roviezzo, F.; Getting, S.J.; Paul-Clark, M.J.; Yona, S.; Gavins, F.N.; Perretti, M.; Hannon, R.; Croxtall, J.D.; Buckingham, J.C.; Flower, R.J. The annexin-1 knockout mouse: What it tells us about the inflammatory response. J. Physiol. Pharmacol. 2002, 53, 541–553. [Google Scholar] [PubMed]
- Petrie, T.A.; Strand, N.S.; Yang, C.T.; Rabinowitz, J.S.; Moon, R.T. Macrophages modulate adult zebrafish tail fin regeneration. Development 2014, 141, 2581–2591. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Chi, M.; Laplace-Builhe, B.; Travnickova, J.; Luz-Crawford, P.; Tejedor, G.; Lutfalla, G.; Kissa, K.; Jorgensen, C.; Djouad, F. TNF signaling and macrophages govern fin regeneration in zebrafish larvae. Cell Death Dis. 2017, 8, e2979. [Google Scholar] [CrossRef]
- Morales, R.A.; Allende, M.L. Peripheral Macrophages Promote Tissue Regeneration in Zebrafish by Fine-Tuning the Inflammatory Response. Front. Immunol. 2019, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Cell Type | Surface Markers | Origin | Maintenance | Functions |
|---|---|---|---|---|
| Cardiac resident macrophages | Ccr2− Timd4+ MHCIIlo | Embryonic | Proliferation in situ >90% |
|
| Cardiac resident macrophages | Ccr2− Timd4− MHCIIhi | Embryonic | Proliferation in situ 75% |
|
| Cardiac resident macrophages | Ccr2+ Timd4− MHCIIhi | Monocytes | Proliferation in situ 15–20% |
|
| Monocytes | Ccr2+ Timd4− MHCIIlo | Monocytes | Monocyte infiltration >99% |
|
| Cell Type | Cluster | Signature Genes | Origin | Functions |
|---|---|---|---|---|
| Cardiac resident macrophages | scRNAseq 1 | Timd4 Folr2 Lyve1 CD163 Igf1 | Embryonic |
|
| Cardiac resident macrophages | scRNAseq 2 | MHC II F4/80 Cx3xr1 CD14 | Embryonic |
|
| Cardiac resident macrophages | scRNAseq 3 | Ccr2 CD64 | Monocytes |
|
| Cardiac resident macrophages | scRNAseq 4 | Ifit1 Ifit3 Irf7 | Monocytes |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez-Argote, S.; O’Meara, C.C. The Evolving Roles of Cardiac Macrophages in Homeostasis, Regeneration, and Repair. Int. J. Mol. Sci. 2021, 22, 7923. https://doi.org/10.3390/ijms22157923
Alvarez-Argote S, O’Meara CC. The Evolving Roles of Cardiac Macrophages in Homeostasis, Regeneration, and Repair. International Journal of Molecular Sciences. 2021; 22(15):7923. https://doi.org/10.3390/ijms22157923
Chicago/Turabian StyleAlvarez-Argote, Santiago, and Caitlin C. O’Meara. 2021. "The Evolving Roles of Cardiac Macrophages in Homeostasis, Regeneration, and Repair" International Journal of Molecular Sciences 22, no. 15: 7923. https://doi.org/10.3390/ijms22157923
APA StyleAlvarez-Argote, S., & O’Meara, C. C. (2021). The Evolving Roles of Cardiac Macrophages in Homeostasis, Regeneration, and Repair. International Journal of Molecular Sciences, 22(15), 7923. https://doi.org/10.3390/ijms22157923