
Microbial Transformation of Galangin Derivatives and Cytotoxicity Evaluation of Their Metabolites

Abstract
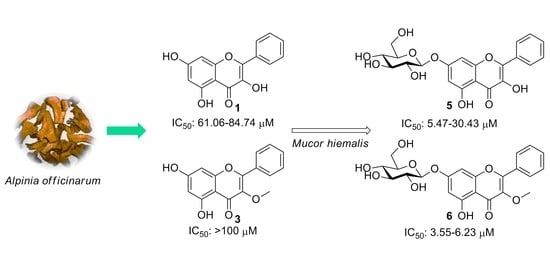
:
1. Introduction
2. Results and Discussion
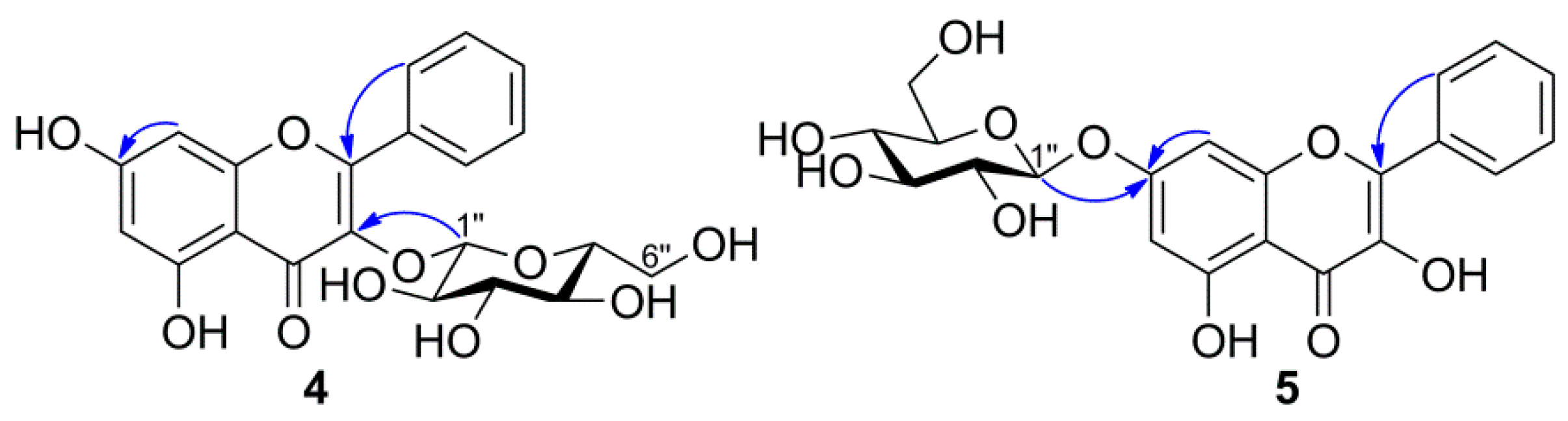
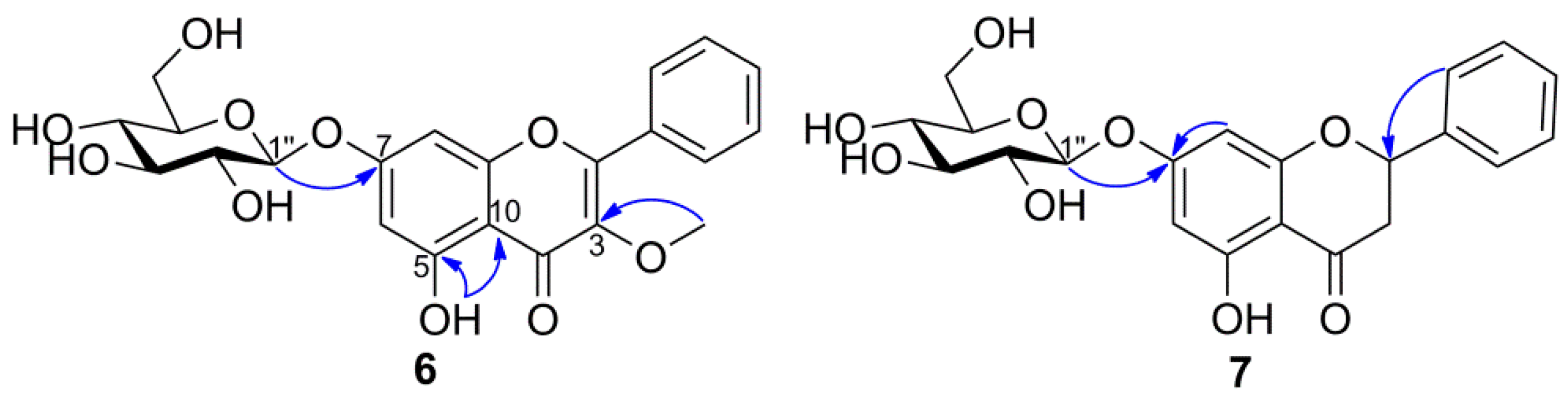
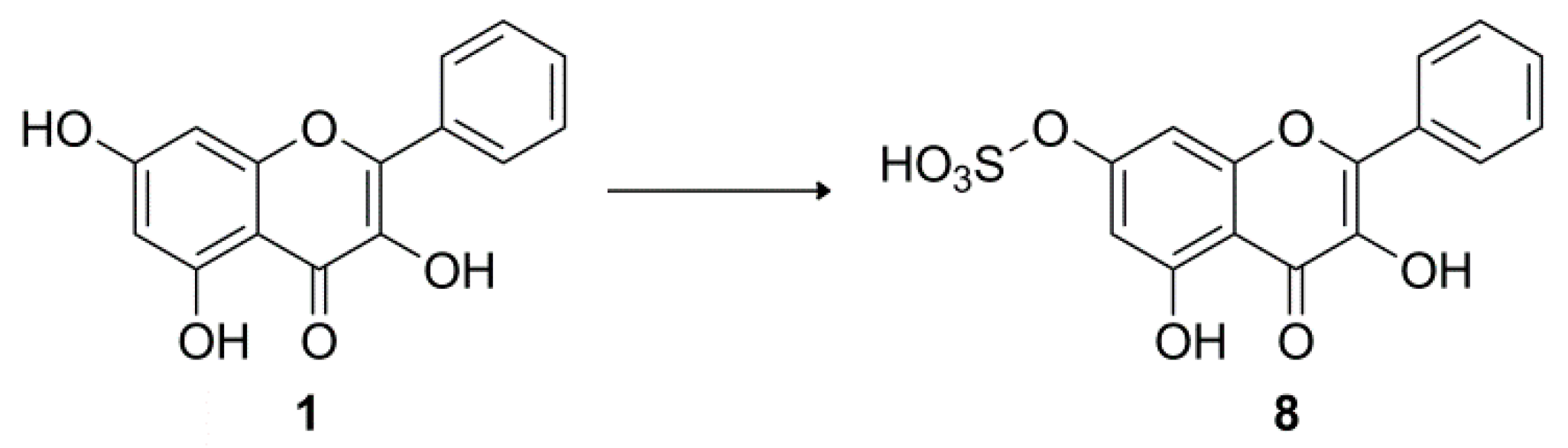
2.1. Identification of Metabolites
2.2. Cytotoxic Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Materials and Microorganisms
3.2.1. Galangin (1)
3.2.2. 3-O-Methylgalangin (2)
3.2.3. Pinocembrin (3)
3.3. Microbial Screening Procedures
3.4. Biotransformation of 1–3 by M. Hiemalis KCTC 26779
3.4.1. Galangin-3-O-β-d-glucopyranoside (4)
3.4.2. Galangin-7-O-β-d-glucopyranoside (5)
3.4.3. 3-O-methylgalangin-7-O-β-d-glucopyranoside (6)
3.4.4. Pinocembrin-7-O-β-d-glucopyranoside (7)
3.5. Biotransformation of 1 by A. coerulea KCTC 6936
Galangin-7-O-sulfate (8)
3.6. Cytotoxic Activity Evaluation
3.7. Hydrolysis of Compounds 4–8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ding, P. Alpinia officinarum Hance (Gaoliangjiang, galangal). In Dietary Chinese Herbs: Chemistry, Pharmacology and Clinical Evidence; Liu, Y., Wang, Z., Zhang, J., Eds.; Springer: Vienna, Austria, 2015; pp. 61–67. [Google Scholar]
- Zhang, J.-Q.; Wang, Y.; Li, H.-L.; Wen, Q.; Yin, H.; Zeng, N.-K.; Lai, W.-Y.; Wei, N.; Cheng, S.-Q.; Kang, S.-L.; et al. Simultaneous quantification of seventeen bioactive components in rhizome and aerial parts of Alpinia officinarum Hance using LC-MS/MS. Anal. Methods 2015, 7, 4919–4926. [Google Scholar] [CrossRef]
- Reid, K.; Wright, V.; Omoregie, S. Anticancer properties of Alpinia officinarum (lesser galangal)-A mini review. Int. J. Adv. Res. 2016, 4, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Leclair, H.M.; Tardif, N.; Paris, A.; Galibert, M.-D.; Corre, S. Role of flavonoids in the prevention of AhR-dependent resistance during treatment with BRAF inhibitors. Int. J. Mol. Sci. 2020, 21, 5025. [Google Scholar] [CrossRef]
- De Oliveira Júnior, R.G.; Ferraz, C.A.A.; Silva, M.G.; de Lavor, É.M.; Rolim, L.A.; de Lima, J.T.; Fleury, A.; Picot, L.; de Souza Siqueira Quintans, J.; Quintans Júnior, L.J.; et al. Flavonoids: Promising natural products for treatment of skin cancer (melanoma). In Natural Products and Cancer Drug Discovery; Badria, F.A., Ed.; IntechOpen: London, UK, 2017; pp. 161–210. [Google Scholar]
- Pillai, M.K.; Young, D.J.; Bin, H.; Abdul Majid, H.M. Therapeutic potential of Alpinia officinarum. Mini Rev. Med. Chem. 2018, 18, 1220–1232. [Google Scholar] [CrossRef]
- Jiao, H.; Xu, S.; Fan, C.; Zhang, Q.; Wang, Y. Chromatographic fingerprint analysis and quantitative evaluate the rhizomes of Alpinia officinarum Hance (lesser galangal). J. Chin. Pharm. Sci. 2019, 28, 728–738. [Google Scholar]
- Basri, A.M.; Taha, H.; Ahmad, N. A review on the pharmacological activities and phytochemicals of Alpinia officinarum (galangal) extracts derived from bioassay-guided fractionation and isolation. Pharmacogn. Rev. 2017, 11, 43–56. [Google Scholar]
- Fang, D.; Xiong, Z.; Xu, J.; Yin, J.; Luo, R. Chemopreventive mechanisms of galangin against hepatocellular carcinoma: A review. Biomed. Pharmacother. 2019, 109, 2054–2061. [Google Scholar] [CrossRef]
- Devadoss, D.; Ramar, M.; Chinnasamy, A. Galangin, a dietary flavonol inhibits tumor initiation during experimental pulmonary tumorigenesis by modulating xenobiotic enzymes and antioxidant status. Arch. Pharm. Res. 2018, 41, 265–275. [Google Scholar] [CrossRef]
- Zhang, W.; Tang, B.; Huang, Q.; Hua, Z. Galangin inhibits tumor growth and metastasis of B16F10 melanoma. J. Cell. Biochem. 2013, 114, 152–161. [Google Scholar] [CrossRef]
- Rasul, A.; Millimouno, F.M.; Eltayb, W.A.; Li, J.; Li, X. Pinocembrin: A novel natural compound with versatile pharmacological and biological activities. Biomed. Res. Int. 2013, 2013, 379850. [Google Scholar] [CrossRef]
- Shen, X.; Liu, Y.; Luo, X.; Yang, Z. Advances in biosynthesis, pharmacology, and pharmacokinetics of pinocembrin, a promising natural small-molecule drug. Molecules 2019, 24, 2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Chen, X.; Li, L.-N.; Tang, W.; Pan, Y.-T.; Kong, J.-Q. Transcriptome-enabled discovery and functional characterization of enzymes related to (2S)-pinocembrin biosynthesis from Ornithogalum caudatum and their application for metabolic engineering. Microb. Cell Fact. 2016, 15, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Wang, K.; Wu, Y.; Chen, Y.; Chen, X.; Hu, C.W.; Hu, F. Pinocembrin induces ER stress mediated apoptosis and suppresses autophagy in melanoma cells. Cancer Lett. 2018, 431, 31–42. [Google Scholar] [CrossRef]
- Khodzhaieva, R.S.; Gladkov, E.S.; Kyrychenko, A.; Roshal, A.D. Progress and achievements in glycosylation of flavonoids. Front. Chem. 2021, 9, 637994. [Google Scholar] [CrossRef] [PubMed]
- Ara, K.Z.G.; Khan, S.; Kulkarni, T.S.; Pozzo, T.; Karlsson, E.N. Glycoside hydrolases for extraction and modification of polyphenolic antioxidants. In Advances in Enzyme Biotechnology; Shukla, P., Pletschke, B.I., Eds.; Springer: New Delhi, India, 2013; pp. 9–21. [Google Scholar]
- Makris, D.P.; Rossiter, J.T. Heat-induced, metal-catalyzed oxidative degradation of quercetin and rutin (quercetin 3-O-rhamnosylglucoside) in aqueous model systems. J. Agric. Food Chem. 2000, 48, 3830–3838. [Google Scholar] [CrossRef] [PubMed]
- Slámová, K.; Kapešová, J.; Valentová, K. “Sweet flavonoids”: Glycosidase-catalyzed modifications. Int. J. Mol. Sci. 2018, 19, 2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruyn, F.D.; Brempt, M.V.; Maertens, J.; Bellegem, W.V.; Duchi, D.; Mey, M.D. Metabolic engineering of Escherichia coli into a versatile glycosylation platform: Production of bioactive quercetin glycosides. Microb. Cell Fact. 2015, 14, 138. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Li, Z.; Wu, Y.; Luo, D.; Qiu, L.; Xie, J.; Li, X. Advances on synthesis of flavonoid glycosides. Chin. J. Org. Chem. 2019, 39, 1875–1890. [Google Scholar] [CrossRef]
- Hayes, M.R.; Pietruszka, J. Synthesis of glycosides by glycosynthases. Molecules 2017, 22, 1434. [Google Scholar] [CrossRef]
- Xu, B.; Liang, G.; Wen, Z.; Hu, Z.; Yuan, J.; Chen, H.; Zhang, L. Synthesis of quercetin glycosides and their α-glucosidase inhibitory activities. Heterocycles 2016, 92, 1245–1260. [Google Scholar]
- Li, Y.F.; Yu, B.; Sun, J.S.; Wang, R.X. Efficient synthesis of baicalin and its analogs. Tetrahedron Lett. 2015, 56, 3816–3819. [Google Scholar] [CrossRef]
- Cano-Flores, A.; Gómez, J.; Escalona-Torres, I.S.; Velasco-Bejarano, B. Microorganisms as biocatalysts and enzyme sources. In Microorganisms; Blumenberg, M., Shaaban, M., Elgaml, A., Eds.; IntechOpen: London, UK, 2020. [Google Scholar]
- Sordon, S.; Popłoński, J.; Huszcza, E. Microbial glycosylation of flavonoids. Pol. J. Microbiol. 2016, 65, 137–151. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Xiao, Y.; Lee, I.-S. Microbial transformation of prenylquercetins by Mucor hiemalis. Molecules 2020, 25, 528. [Google Scholar] [CrossRef] [Green Version]
- Lipkind, G.M.; Shashkov, A.S.; Knirel, Y.A.; Vinogradov, E.V.; Kochetkov, N.K. A computer-assisted structural analysis of regular polysaccharides on the basis of 13C-n.m.r. data. Carbohyr. Res. 1988, 175, 59–75. [Google Scholar] [CrossRef]
- Jiang, J.; Fang, S.; Xu, C.; Luo, J. Studies on the chemical constituents of Rourea microphylla (Hook. et Arn) Planch. Acta Bot. Sin. 1990, 32, 376–379. [Google Scholar]
- Pyo, M.K.; Yun-Choi, H.S.; Kim, Y.K. Isolation of n-butyl-β-D-fructopyranoside from Gastrodia elata Blume. Nat. Prod. Sci. 2006, 12, 101–103. [Google Scholar]
- Liu, G.; Sun, L.; Wang, S.; Chen, C.; Guo, T.; Ji, Y.; Li, X.; Huang, G.; Wei, H.; Dai, Y.; et al. Hydroxylation modification and free radical scavenging activity of puerarin-7-O-fructoside. Folia Microbiol. 2011, 56, 305–311. [Google Scholar] [CrossRef]
- Esaki, S.; Abe, M.; Shiba, N.; Kamiya, S. Synthesis of glycyphyllin. Agric. Biol. Chem. 1990, 54, 1583–1585. [Google Scholar]
- Zapesochnaya, G.G. Datisca cannabina flavonoids. VII. Galanginin–a new glycoside of galangin. Khim. Prir. Soedin. 1982, 5, 651. [Google Scholar]
- Antri, A.E.; Messouri, I.; Tlemçani, R.C.; Bouktaib, M.; El Alami, R.; El Bali, B.; Lachkar, M. Flavone glycosides from Calycotome villosa Subsp. Intermedia Mol. 2004, 9, 568–573. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhao, J.; Guo, J.; Mei, S.; Li, L. Flavone compounds isolated from Lagotis Yunnanensis. Chin. Tradit. Herb. Drugs 2004, 35, 257–259. [Google Scholar]
- Feng, H.; Wang, Z.; Dong, G.; Wu, Z. Studies on chemical constituents from Penthorum chinense Pursh. Zhongguo Zhongyao Zazhi 2001, 26, 260–262. [Google Scholar]
- Varin, L. Flavonoid sulfation: Phytochemistry, enzymology and molecular biology. In Phenolic Metabolism in Plants. Recent Advances in Phytochemistry; Stafford, H.A., Ibrahim, R.K., Eds.; Plenum Press: New York, NY, USA; Springer: Boston, MA, USA, 1992; Volume 26, pp. 233–254. [Google Scholar]
- Ibrahim, A.R.S. Sulfation of naringenin by Cunninghamella elegans. Phytochemistry 2000, 53, 209–212. [Google Scholar] [CrossRef]
- Teles, Y.C.F.; Souza, M.S.R.; de Souza, M.F.V. Sulphated flavonoids: Biosynthesis, structures, and biological activities. Molecules 2018, 23, 480. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Xiao, Y.; Lee, I.-S. Microbial conjugation studies of licochalcones and xanthohumol. Int. J. Mol. Sci. 2021, 22, 6893. [Google Scholar] [CrossRef]
- Enerstvedt, K.H.; Jordheim, M.; Andersen, Ø.M. Isolation and identification of flavonoids found in Zostera marina collected in Norwegian coastal waters. Am. J. Plant. Sci. 2016, 7, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- An, N.; Yang, S.; Zou, Z.; Xu, L. Flavonoids of Alpinia officinarum. Chin. Tradit. Herb Drugs 2006, 37, 663–664. [Google Scholar]
- Goodarzi, S.; Tabatabaei, M.J.; Jafari, R.M.; Shemirani, F.; Tavakoli, S.; Mofasseri, M.; Tofighi, Z. Cuminum cyminum fruits as source of luteolin-7-O-glucoside, potent cytotoxic flavonoid against breast cancer cell lines. Nat. Prod. Res. 2020, 34, 1602–1606. [Google Scholar] [CrossRef] [PubMed]
- Smiljkovic, M.; Stanisavljevic, D.; Stojkovic, D.; Petrovic, I.; Vicentic, J.M.; Popovic, J.; Grdadolnik, S.G.; Markovic, D.; Sankovic-Babice, S.; Glamoclija, J.; et al. Apigenin-7-O-glucoside versus apigenin: Insight into the modes of anticandidal and cytotoxic actions. Excli J. 2017, 16, 795–807. [Google Scholar] [PubMed]
- Ikemoto, S.; Sugimura, K.; Yoshida, N.; Yasumoto, R.; Wada, S.; Yamamoto, K.; Kishimoto, T. Antitumor effects of Scutellariae radix and its components baicalein, baicalin, wogonin on bladder cancer cell lines. Urology 2000, 55, 951–955. [Google Scholar] [CrossRef]
- Aisyah, L.S.; Yun, Y.F.; Herlina, T.; Julaeha, E.; Zainuddin, A.; Nurfarida, I.; Hidayat, A.T.; Supratman, U.; Shiono, Y. Flavonoid compounds from the leaves of Kalanchoe prolifera and their cytotoxic activity against P-388 murine leukimia cells. Nat. Prod. Sci. 2017, 23, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Jing, N.; Song, J.; Liu, Z.; Wang, L.; Jiang, G. Glycosylation of anthocyanins enhances the apoptosis of colon cancer cells by handicapping energy metabolism. BMC Complement. Med. Ther. 2020, 20, 312. [Google Scholar] [CrossRef]
- Zou, T.B.; Feng, D.; Song, G.; Li, H.W.; Tang, H.W.; Ling, W.H. The role of sodium-dependent glucose transporter 1 and glucose transporter 2 in the absorption of cyanidin-3-O-β-glucoside in Caco-2 cells. Nutrients 2014, 6, 4165–4177. [Google Scholar] [CrossRef] [PubMed]
- Walton, M.C.; McGhie, T.K.; Reynolds, G.W.; Hendriks, W.H. The flavonol quercetin-3-glucoside inhibits cyanidin-3-glucoside absorption in vitro. J. Agric. Food Chem. 2006, 54, 4913–4920. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.A.; Owen, G.I. Glucose transporters: Expression, regulation and cancer. Biol. Res. 2002, 35, 9–26. [Google Scholar] [CrossRef]
- Laudański, P.; Swiatecka, J.; Kovalchuk, O.; Wołczyński, S. Expression of GLUT1 gene in breast cancer cell lines MCF-7 and MDA-MB-231. Ginekol. Pol. 2003, 74, 782–785. [Google Scholar]
- Koch, A.; Lang, S.A.; Wild, P.J.; Gantner, S.; Mahli, A.; Spanier, G.; Berneburg, M.; Müller, M.; Bosserhoff, A.K.; Hellerbrand, C. Glucose transporter isoform 1 expression enhances metastasis of malignant melanoma cells. Oncotarget 2015, 6, 32748–32760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J. Dietary flavonoid aglycones and their glycosides: Which show better biological significance? Crit. Rev. Food Sci. Nutr. 2017, 57, 1874–1905. [Google Scholar] [CrossRef]
- Srivastava, J.K.; Gupta, S. Extraction, characterization, stability and biological activity of flavonoids isolated from chamomile flowers. Mol. Cell Pharmacol. 2009, 1, 138–147. [Google Scholar] [CrossRef]
- Amador, S.; Nieto-Camacho, A.; Ramírez-Apan, M.T.; Martínez, M.; Maldonado, E. Cytotoxic, anti-inflammatory, and α-glucosidase inhibitory effects of flavonoids from Lippia graveolens (Mexican oregano). Med. Chem. Res. 2020, 29, 1497–1506. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Han, F.; Lee, I.-S. A new flavonol glycoside from the aerial parts of Epimedium koreanum Nakai. Nat. Prod. Res. 2017, 33, 320–325. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Cell Lines (IC50, μM) | ||||
|---|---|---|---|---|---|
| MCF-7 | A375P | B16F10 | B16F1 | A549 | |
| 1 | 79.44 ± 2.53 | 84.74 ± 1.70 | 61.06 ± 2.50 | 62.46 ± 4.81 | 63.64 ± 6.24 |
| 2 | 99.83 ± 4.13 | >100 | >100 | >100 | >100 |
| 3 | 66.56 ± 0.45 | 55.35 ± 0.85 | 24.02 ± 0.77 | 30.15 ± 1.85 | 48.33 ± 1.56 |
| 4 | >100 | >100 | >100 | >100 | >100 |
| 5 | 5.47 ± 2.16 | 30.43 ± 0.69 | 12.65 ± 0.74 | 28.65 ± 2.75 | 6.30 ± 0.34 |
| 6 | 3.55 ± 0.49 | 6.23 ± 1.95 | 4.38 ± 0.06 | 3.75 ± 0.25 | 4.83 ± 0.90 |
| 7 | 104.30 ± 2.70 | 110.60 ± 1.50 | 62.43 ± 1.79 | 80.62 ± 5.46 | 39.34 ± 1.89 |
| DZ 1 | 5.12 ± 0.44 | 9.86 ± 0.57 | 6.61 ± 0.83 | 5.42 ± 0.46 | 4.01 ± 0.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, F.; Xiao, Y.; Lee, I.-S. Microbial Transformation of Galangin Derivatives and Cytotoxicity Evaluation of Their Metabolites. Catalysts 2021, 11, 1020. https://doi.org/10.3390/catal11091020
Han F, Xiao Y, Lee I-S. Microbial Transformation of Galangin Derivatives and Cytotoxicity Evaluation of Their Metabolites. Catalysts. 2021; 11(9):1020. https://doi.org/10.3390/catal11091020
Chicago/Turabian StyleHan, Fubo, Yina Xiao, and Ik-Soo Lee. 2021. "Microbial Transformation of Galangin Derivatives and Cytotoxicity Evaluation of Their Metabolites" Catalysts 11, no. 9: 1020. https://doi.org/10.3390/catal11091020