
The Effects of Fibrinogen’s Interactions with Its Neuronal Receptors, Intercellular Adhesion Molecule-1 and Cellular Prion Protein


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Setups and Groups
2.2. Cell Culture
2.3. Materials and Reagents
2.4. Proximity Ligation Assay (PLA)
2.5. Quantitative Real-Time PCR (qRT-PCR)
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. ROS, MitoSOX and Nitrite Detection
2.8. LIVE/DEAD® Viability/Cytotoxicity Assay
2.9. Image Analysis
2.10. Statistical Analysis
3. Results
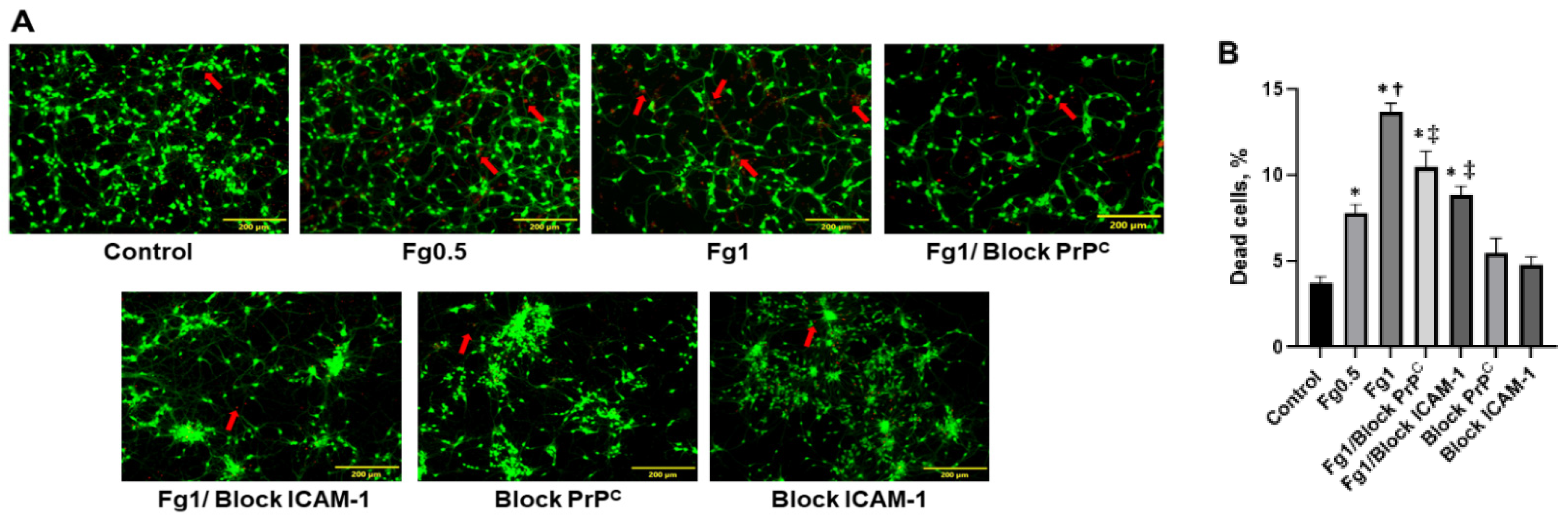
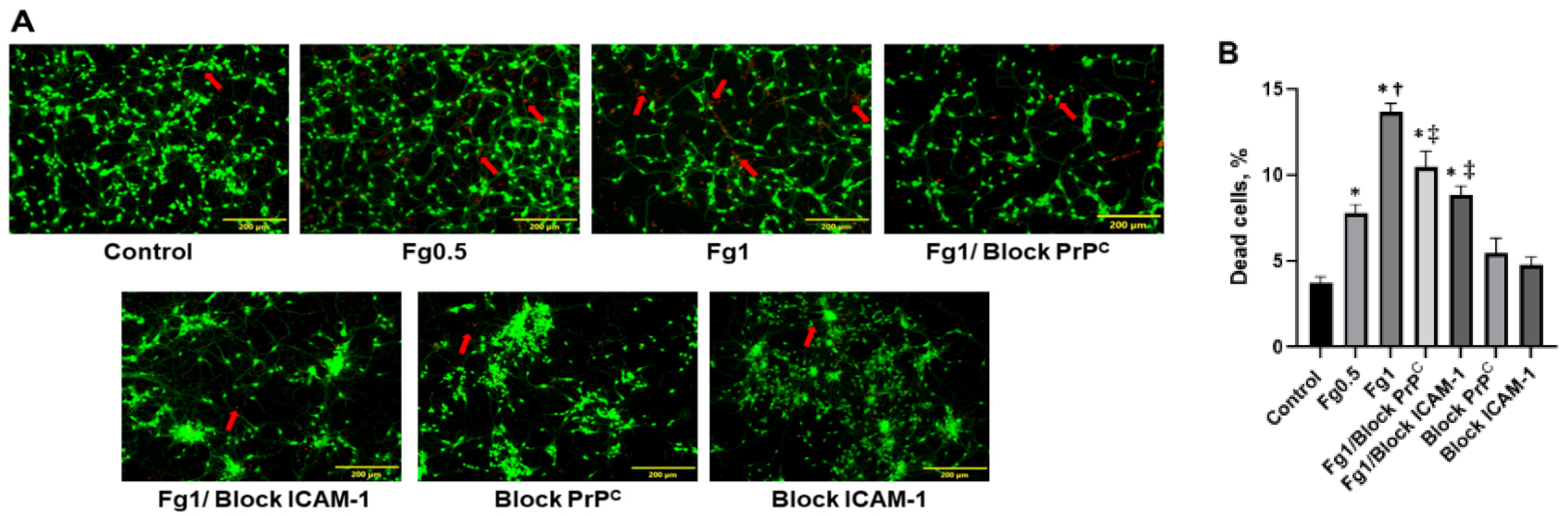
3.1. Fg Increased Death of Neurons
3.2. The Association of Fg with PrPC and ICAM-1 on the Surface of Neurons
3.3. Fg-Induced Upregulation of IL-6 in Neurons
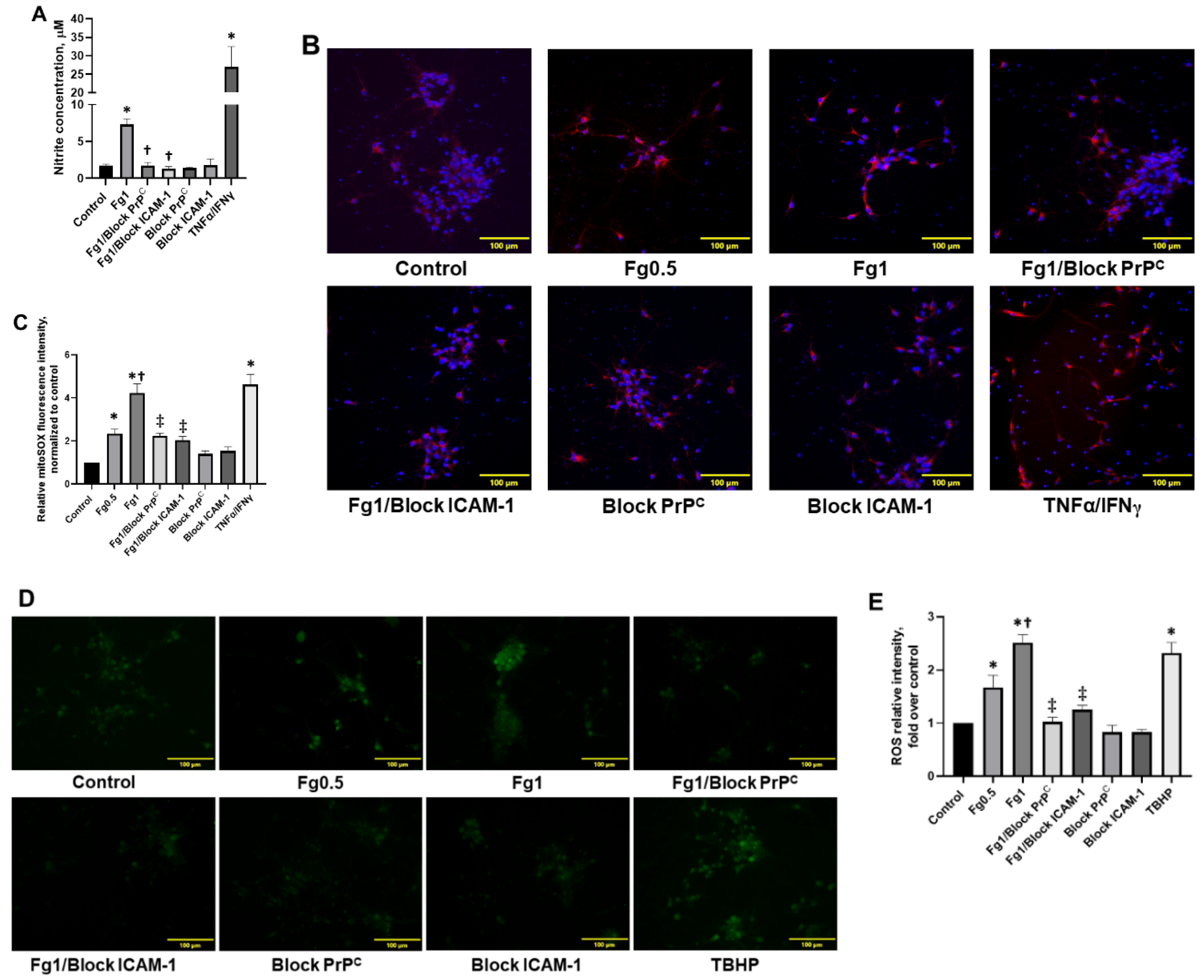
3.4. Fg-Induced Oxidative Stress in Neurons
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| TBI | traumatic brain injury |
| PrPC | cellular prion protein |
| STM | short-term memory |
| ICAM-1 | intercellular adhesion molecule-1 |
| IL-6 | interleukin 6 |
| ROS | reactive oxygen species |
| Fg | fibrinogen |
| m-mTBI | mild-to-moderate traumatic brain injury |
| TNFα | Tumor Necrosis Factor alpha |
| IFNγ | interferon gamma |
| PLA | proximity ligation assay |
| IgG | Immunoglobulin G |
| ELISA | enzyme-linked immunosorbent assay |
| qRT-PCR | quantitative real-time PCR |
| TBHP | tert-butyl hydroperoxide |
| MS | multiple sclerosis |
| CSF | cerebrospinal fluid |
| NAPDH | dinucleotide phosphate oxidase |
| EthD-1 | ethidium homodimer-1 |
| AOI | area of interests |
| ANOVA | analysis of variance |
References
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef]
- Hay, J.R.; Johnson, V.E.; Young, A.M.; Smith, D.H.; Stewart, W. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar]
- Muradashvili, N.; Benton, R.L.; Saatman, K.E.; Tyagi, S.C.; Lominadze, D. Ablation of matrix metalloproteinase-9 gene decreases cerebrovascular permeability and fibrinogen deposition post traumatic brain injury in mice. Metab. Brain Dis. 2015, 30, 411–426. [Google Scholar] [CrossRef] [Green Version]
- Muradashvili, N.; Tyagi, S.C.; Lominadze, D. Localization of fibrinogen in the vasculo-astrocyte interface after cortical contusion injury in mice. Brain Sci. 2017, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muradashvili, N.; Charkviani, M.; Sulimai, N.; Tyagi, N.; Crosby, J.; Lominadze, D. Effects of fibrinogen synthesis inhibition on vascular cognitive impairment during traumatic brain injury in mice. Brain Res. 2020, 1751, 147208. [Google Scholar] [CrossRef] [PubMed]
- Marchant, R.E.; Kang, I.; Sit, P.S.; Zhou, Y.; Todd, B.A.; Eppell, S.J.; Lee, I. Molecular views and measurements of hemostatic processes using atomic force microscopy. Curr. Protein Pept. Sci. 2002, 3, 249–274. [Google Scholar] [CrossRef] [Green Version]
- Potschka, M. Universal calibration of gel permeation chromatography and determination of molecular shape in solution. Anal. Biochem. 1987, 162, 47–64. [Google Scholar] [CrossRef]
- Fu, B.M.; Shen, S. Structural mechanisms of acute VEGF effect on microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H2124–H2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tennent, G.A.; Brennan, S.O.; Stangou, A.J.; O’Grady, J.; Hawkins, P.N.; Pepys, M.B. Human plasma fibrinogen is synthesized in the liver. Blood 2007, 109, 1971–1974. [Google Scholar] [CrossRef]
- Muradashvili, N.; Tyagi, R.; Tyagi, N.; Tyagi, S.C.; Lominadze, D. Cerebrovascular disorders caused by hyperfibrinogenemia. J. Physiol. 2016, 594, 5941–5957. [Google Scholar] [CrossRef]
- Muradashvili, N.; Khundmiri, S.J.; Tyagi, R.; Gartung, A.; Dean, W.L.; Lee, M.-J.; Lominadze, D. Sphingolipids affect fibrinogen-induced caveolar transcytosis and cerebrovascular permeability. Am. J. Physiol. Cell Physiol. 2014, 307, C169–C179. [Google Scholar] [CrossRef] [Green Version]
- Muradashvili, N.; Tyagi, R.; Lominadze, D. A dual-tracer method for differentiating transendothelial transport from paracellular leakage in vivo and in vitro. Front. Physiol. 2012, 3, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrP(C)): Its physiological function and role in disease. Biochim. Biophys. Acta 2007, 1772, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Scholz, D.; Devaux, B.; Hirche, A.; Pötzsch, B.; Kropp, B.; Schaper, W.; Schaper, J. Expression of adhesion molecules is specific and time-dependent in cytokine-stimulated endothelial cells in culture. Cell Tissue Res. 1996, 284, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Wang, J.G.; Zhang, J.Z.; Cao, C.X.; Chang, Y.; Dong, J.D.; Guo, F.Y.; Li, P.A. Upregulation of ICAM-1 in diabetic rats after transient forebrain ischemia and reperfusion injury. J. Inflamm. 2014, 11, 35. [Google Scholar] [CrossRef] [Green Version]
- Héry, C.; Sébire, G.; Peudenier, S.; Tardieu, M. Adhesion to human neurons and astrocytes of monocytes: The role of interaction of CR3 and ICAM-1 and modulation by cytokines. J. Neuroimmunol. 1995, 57, 101–109. [Google Scholar] [CrossRef]
- Charkviani, M.; Muradashvili, N.; Sulimai, N.H.; Lominadze, D. Fibrinogen—Cellular prion protein complex formation on astrocytes. J. Neurophysiol. 2020, 124, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Languino, L.R.; Plescia, J.; Duperrray, A.; Brian, A.A.; Plow, E.F.; Geltosky, J.E.; Alteri, D.C. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell 1993, 73, 1423–1434. [Google Scholar] [CrossRef]
- Harley, S.L.; Powell, J.T. Interaction of fibrinogen with saphenous vein endothelial cells stimulates tyrosine phosphorylation of cortactin. Endothelium 2000, 7, 149–154. [Google Scholar] [CrossRef]
- Hubbard, A.K.; Rothlein, R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free. Radic. Biol. Med. 2000, 28, 1379–1386. [Google Scholar] [CrossRef]
- Harley, S.L.; Sturge, J.; Powell, J.T. Regulation by fibrinogen and its products of intercellular adhesion molecule-1 expression in human saphenous vein endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Canteli, M.; Paul, J.; Norris, E.H.; Bronstein, R.; Ahn, H.J.; Zamolodchikov, D.; Bhuvanendran, S.; Fenz, K.M.; Strickland, S. Fibrinogen and β-Amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 2010, 66, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Onodera, T. Dual role of cellular prion protein in normal host and Alzheimer’s disease. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 155–173. [Google Scholar] [CrossRef] [Green Version]
- Sulimai, N.; Brown, J.; Lominadze, D. Fibrinogen interaction with astrocyte ICAM-1 and PrPC results in the generation of ROS and neuronal death. Int. J. Mol. Sci. 2021, 22, 2391. [Google Scholar] [CrossRef]
- Clark, V.D.; Layson, A.; Charkviani, M.; Muradashvili, N.; Lominadze, D. Hyperfibrinogenemia-mediated astrocyte activation. Brain Res. 2018, 1699, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Miklossy, J.; Klegeris, A.; Guo, J.-P.; McGeer, P.L. Thrombin and prothrombin are expressed by neurons and glial cells and accumulate in neurofibrillary tangles in Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 2006, 65, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, M.; Kauffman, M.; Zhu, H.; Trush, M.; Jia, Z.; Li, R. MitoSOX-based flow cytometry for detecting mitochondrial ROS. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lominadze, D.; Tsakadze, N.; Sen, U.; Falcone, J.C.; D’Souza, S.E. Fibrinogen- and fragment D-induced vascular constriction. Am. J. Physiol. 2005, 288, H1257–H1264. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1078, pp. 9–21. [Google Scholar]
- Yates, R.L.; Esiri, M.M.; Palace, J.; Jacobs, B.; Perera, R.; DeLuca, G.C. Fibrin(ogen) and neurodegeneration in the progressive multiple sclerosis cortex. Ann. Neurol. 2017, 82, 259–270. [Google Scholar] [CrossRef]
- Jenkins, D.R.; Craner, M.J.; Esiri, M.M.; DeLuca, G.C. The contribution of fibrinogen to inflammation and neuronal density in human traumatic brain injury. J. Neurotrauma 2018, 35, 2259–2271. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, J.; Wu, X.; Xi, C.; Gai, Y.; Liu, H.; Yuan, Q.; Wang, E.; Gao, L.; Hu, J.; et al. Validating the incidence of coagulopathy and disseminated intravascular coagulation in patients with traumatic brain injury—Analysis of 242 cases. Br. J. Neurosurg. 2011, 25, 363–368. [Google Scholar] [CrossRef]
- Fiala, M.; Liu, Q.N.; Sayre, J.; Pop, V.; Brahmandam, V.; Graves, M.C.; Vinters, H.V. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood–brain barrier. Eur. J. Clin. Investig. 2002, 32, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.J.; Zamolodchikov, D.; Cortes-Canteli, M.; Norris, E.H.; Glickman, J.F.; Strickland, S. Alzheimer’s disease peptide β-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. USA 2010, 107, 21812–21817. [Google Scholar] [CrossRef] [Green Version]
- Vos, C.M.P.; Geurts, J.J.G.; Montagne, L.; van Haastert, E.S.; Bö, L.; van der Valk, P.; Barkhof, F.; de Vries, H.E. Blood–brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiol. Dis. 2005, 20, 953–960. [Google Scholar] [CrossRef]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-[beta] pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.; Kim, J.-H.; Lee, S.; Kim, J.-H.; Seo, J.-W.; Jin, M.; Lee, M.-G.; Jang, I.-S.; Lee, W.-H.; Suk, K. Phenotypic polarization of activated astrocytes: The critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. J. Immunol. 2013, 191, 5204. [Google Scholar] [CrossRef] [Green Version]
- Tarassishin, L.; Suh, H.-S.; Lee, S.C. LPS and IL-1 differentially activate mouse and human astrocytes: Role of CD14. Glia 2014, 62, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.P.Q.; Perreau, V.M.; Shultz, S.R.; Brady, R.D.; Lei, E.; Dixit, S.; Taylor, J.M.; Beart, P.M.; Boon, W.C. Inflammation in traumatic brain injury: Roles for toxic A1 astrocytes and microglial-astrocytic crosstalk. Neurochem. Res. 2019, 44, 1410–1424. [Google Scholar] [CrossRef]
- Morganti-Kossman, M.C.; Lenzlinger, P.M.; Hans, V.; Stahel, P.; Csuka, E.; Ammann, E.; Stocker, R.; Trentz, O.; Kossmann, T. Production of cytokines following brain injury: Beneficial and deleterious for the damaged tissue. Mol. Psychiatry 1997, 2, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Kossmann, T.; Hans, V.H.; Imhof, H.G.; Stocker, R.; Grob, P.; Trentz, O.; Morganti-Kossmann, C. Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock 1995, 4, 311–317. [Google Scholar] [CrossRef]
- Harting, M.T.; Jimenez, F.; Adams, S.D.; Mercer, D.W.; Cox, C.S., Jr. Acute, regional inflammatory response after traumatic brain injury: Implications for cellular therapy. Surgery 2008, 144, 803–813. [Google Scholar] [CrossRef] [Green Version]
- Avery, S. Oxidative stress and cell function. Syst. Biol. Free Radic. Antioxid. 2012, 89–112. [Google Scholar] [CrossRef]
- Batandier, C.; Fontaine, E.; Kériel, C.; Leverve, X.M. Determination of mitochondrial reactive oxygen species: Methodological aspects. J. Cell. Mol. Med. 2002, 6, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, J.K.; Rafalski, V.A.; Meyer-Franke, A.; Adams, R.A.; Poda, S.B.; Rios Coronado, P.E.; Pedersen, L.Ø.; Menon, V.; Baeten, K.M.; Sikorski, S.L.; et al. Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 2018, 19, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Kyu Ryu, J.; Merlini, M.; Baeten, K.M.; Le Moan, N.; Petersen, M.A.; Deerinck, T.J.; Smirnoff, D.S.; Bedard, C.; Hakozaki, H.; et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 2012, 3, 1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlini, M.; Rafalski, V.A.; Rios Coronado, P.E.; Gill, T.M.; Ellisman, M.; Muthukumar, G.; Subramanian, K.S.; Ryu, J.K.; Syme, C.A.; Davalos, D.; et al. Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer’s disease model. Neuron 2019, 101, 1099–1108.e1096. [Google Scholar] [CrossRef] [Green Version]
- Perez, R.L.; Roman, J. Fibrin enhances the expression of IL-1 beta by human peripheral blood mononuclear cells. Implications in pulmonary inflammation. J. Immunol. 1995, 154, 1879. [Google Scholar] [PubMed]
- Schneider, B.; Mutel, V.; Mathéa Pietri, M.; Ermonval, M.; Mouillet-Richard, S.; Kellermann, O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc.Natl. Acad. Sci. USA 2003, 100, 13326–13331. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulimai, N.; Brown, J.; Lominadze, D. The Effects of Fibrinogen’s Interactions with Its Neuronal Receptors, Intercellular Adhesion Molecule-1 and Cellular Prion Protein. Biomolecules 2021, 11, 1381. https://doi.org/10.3390/biom11091381
Sulimai N, Brown J, Lominadze D. The Effects of Fibrinogen’s Interactions with Its Neuronal Receptors, Intercellular Adhesion Molecule-1 and Cellular Prion Protein. Biomolecules. 2021; 11(9):1381. https://doi.org/10.3390/biom11091381
Chicago/Turabian StyleSulimai, Nurul, Jason Brown, and David Lominadze. 2021. "The Effects of Fibrinogen’s Interactions with Its Neuronal Receptors, Intercellular Adhesion Molecule-1 and Cellular Prion Protein" Biomolecules 11, no. 9: 1381. https://doi.org/10.3390/biom11091381