
An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy

 ,
,  and
and 
Abstract
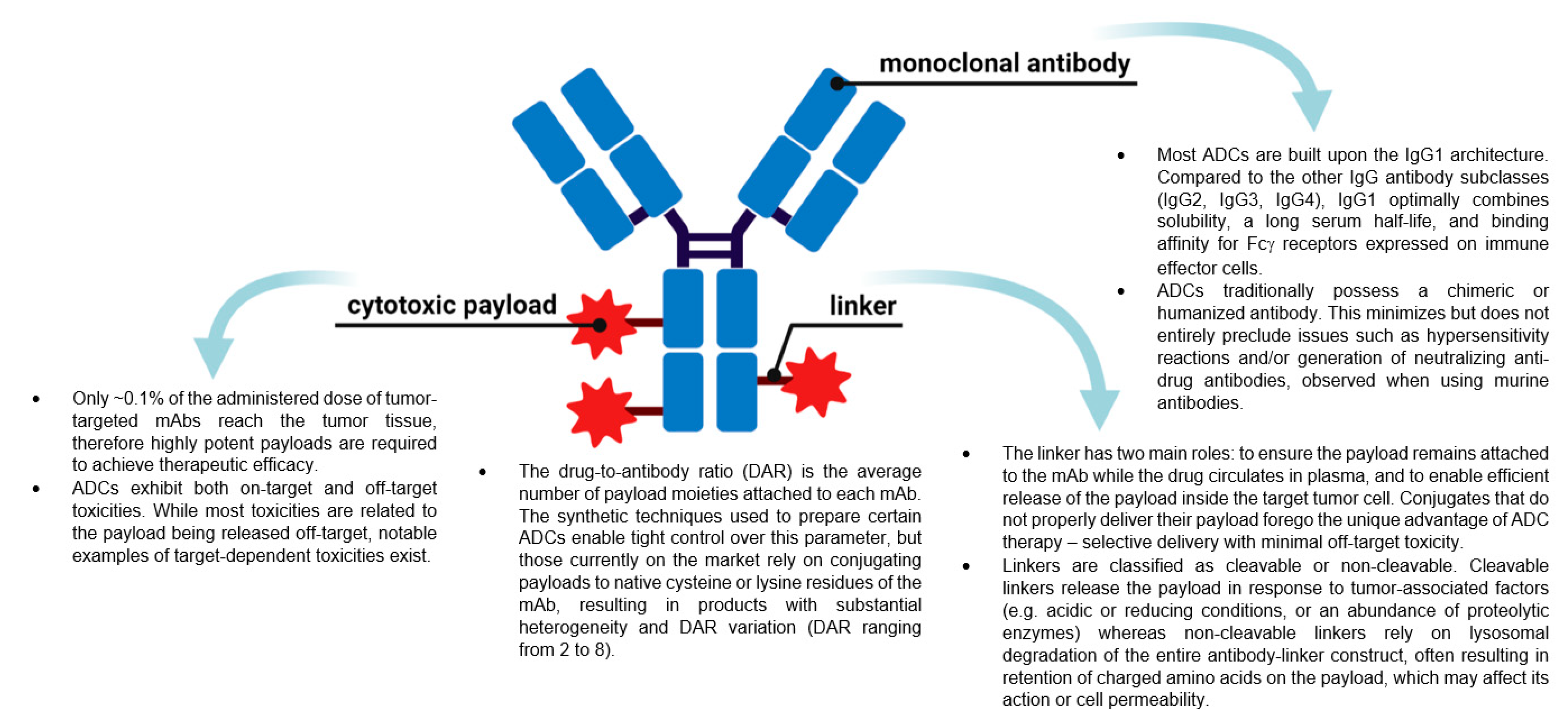
:1. Introduction
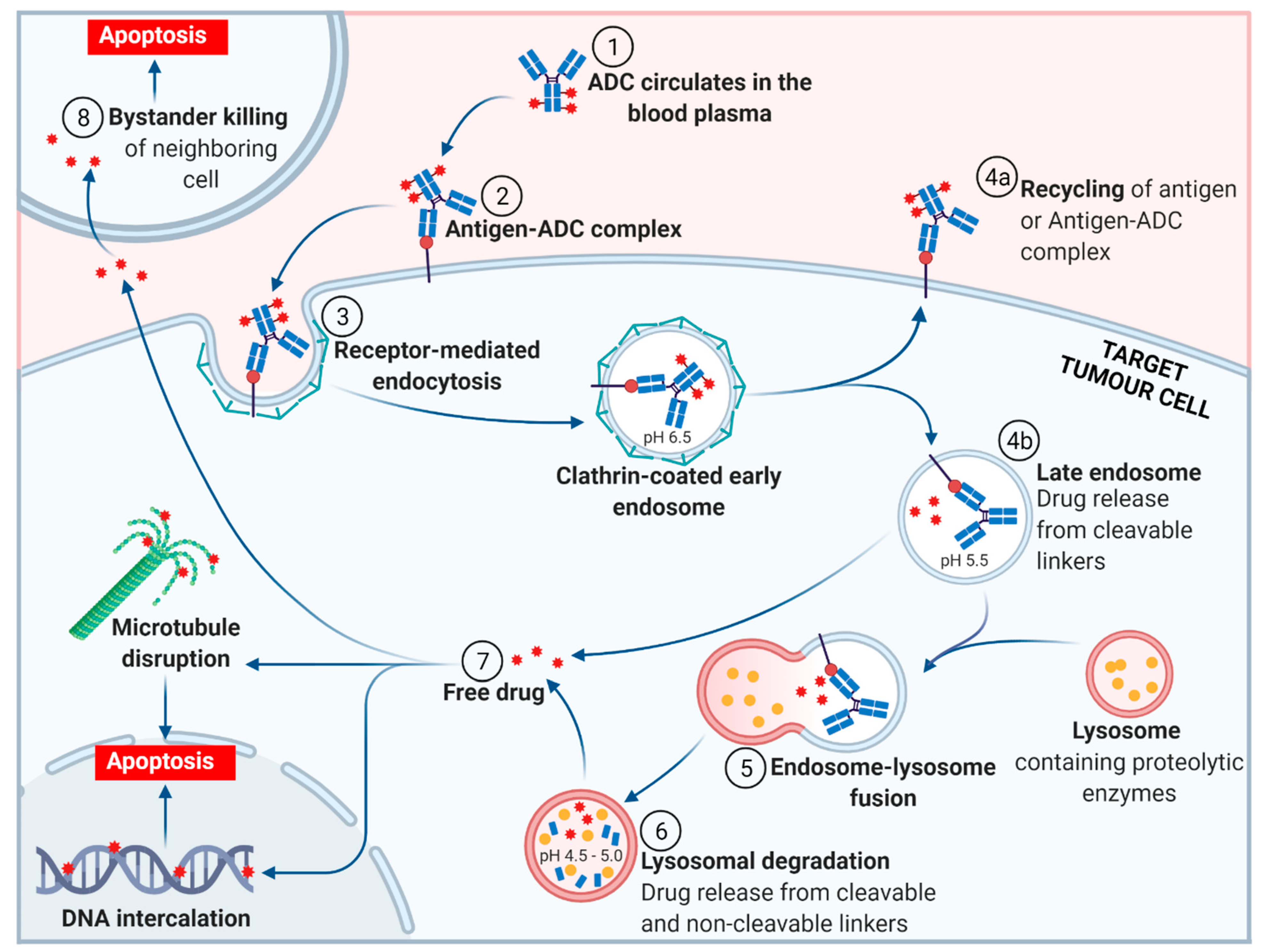
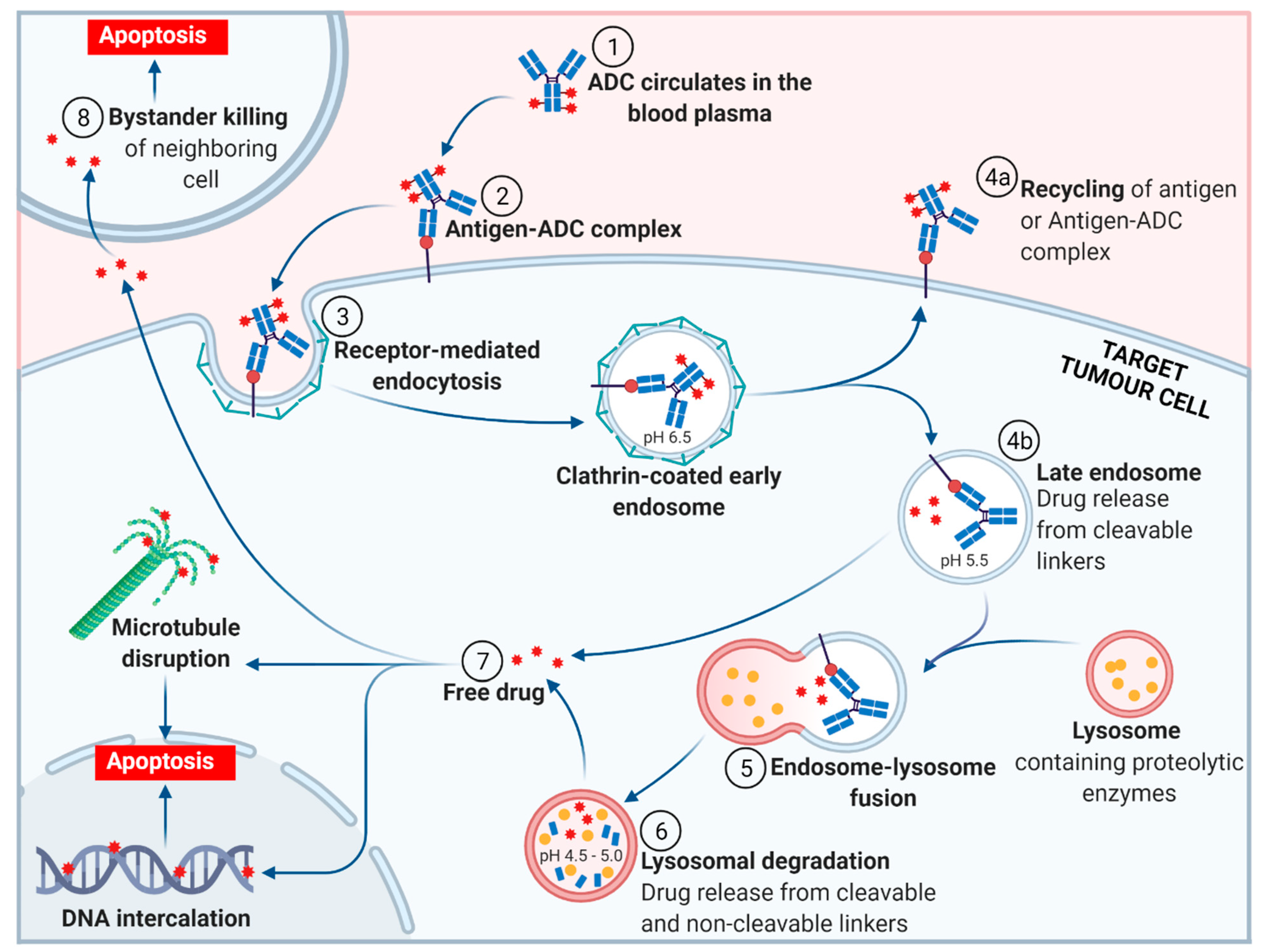
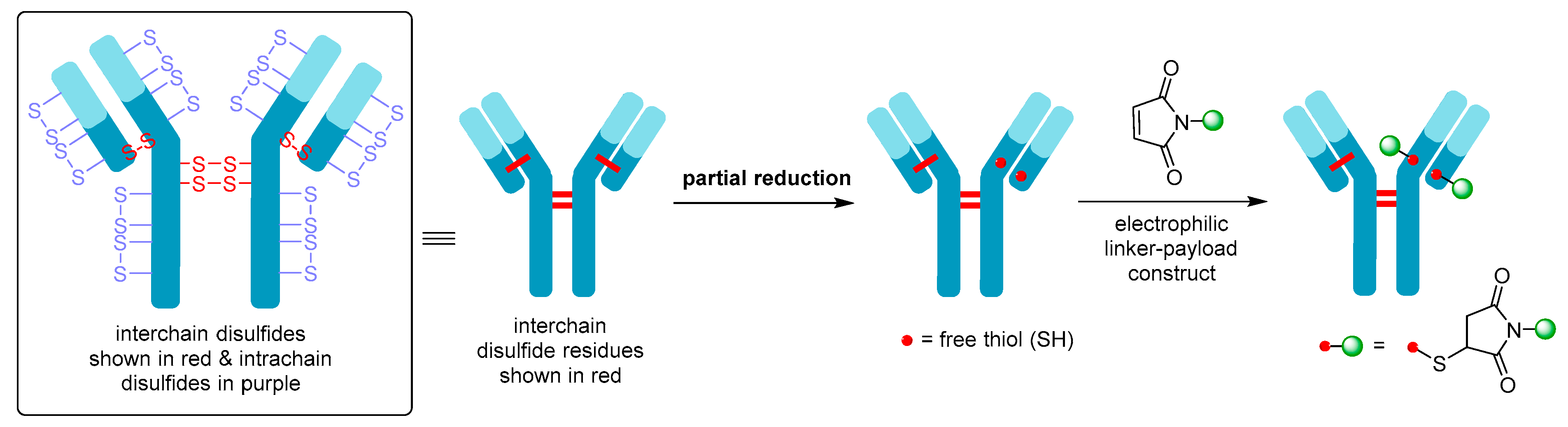
2. ADC Mechanism of Action
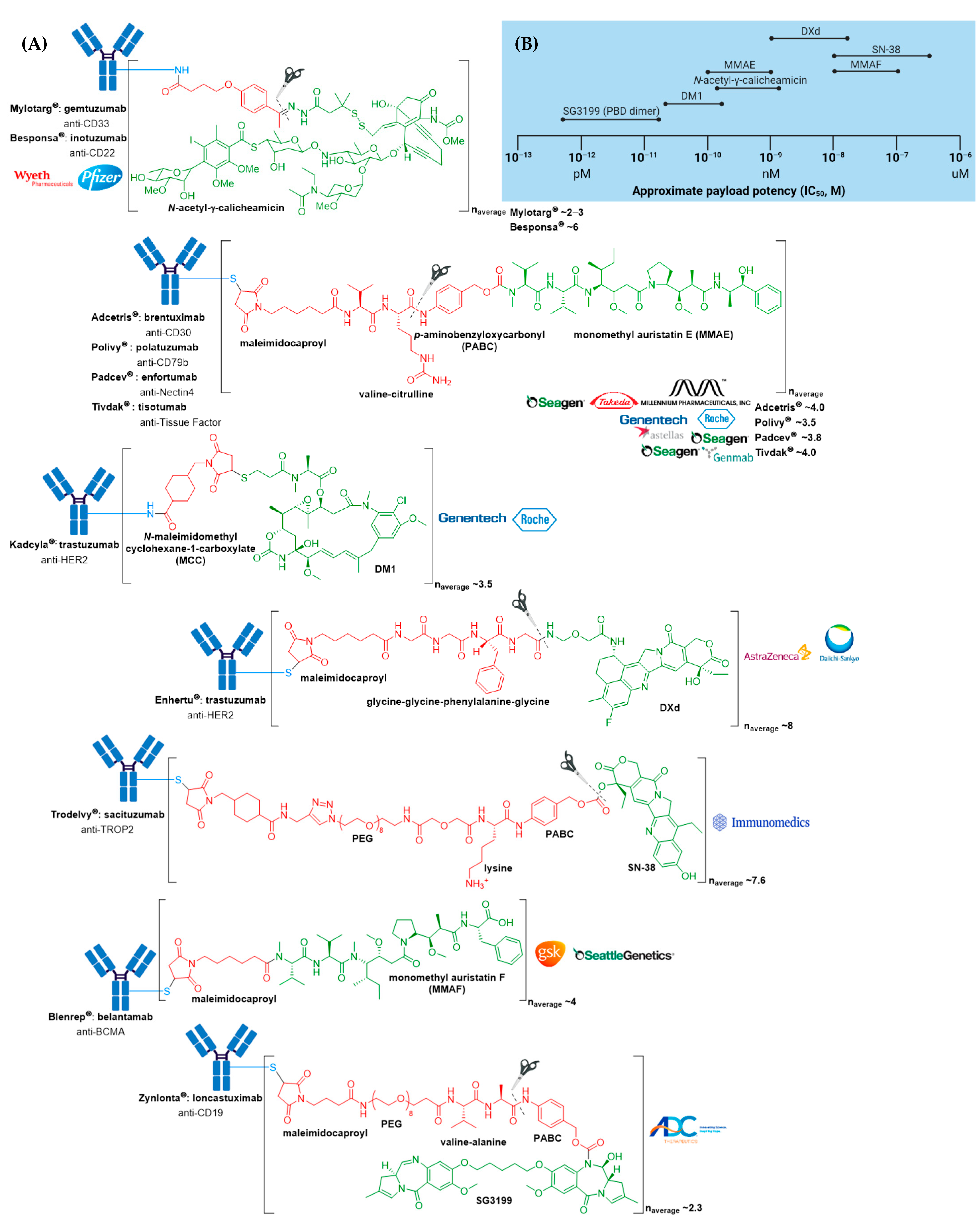
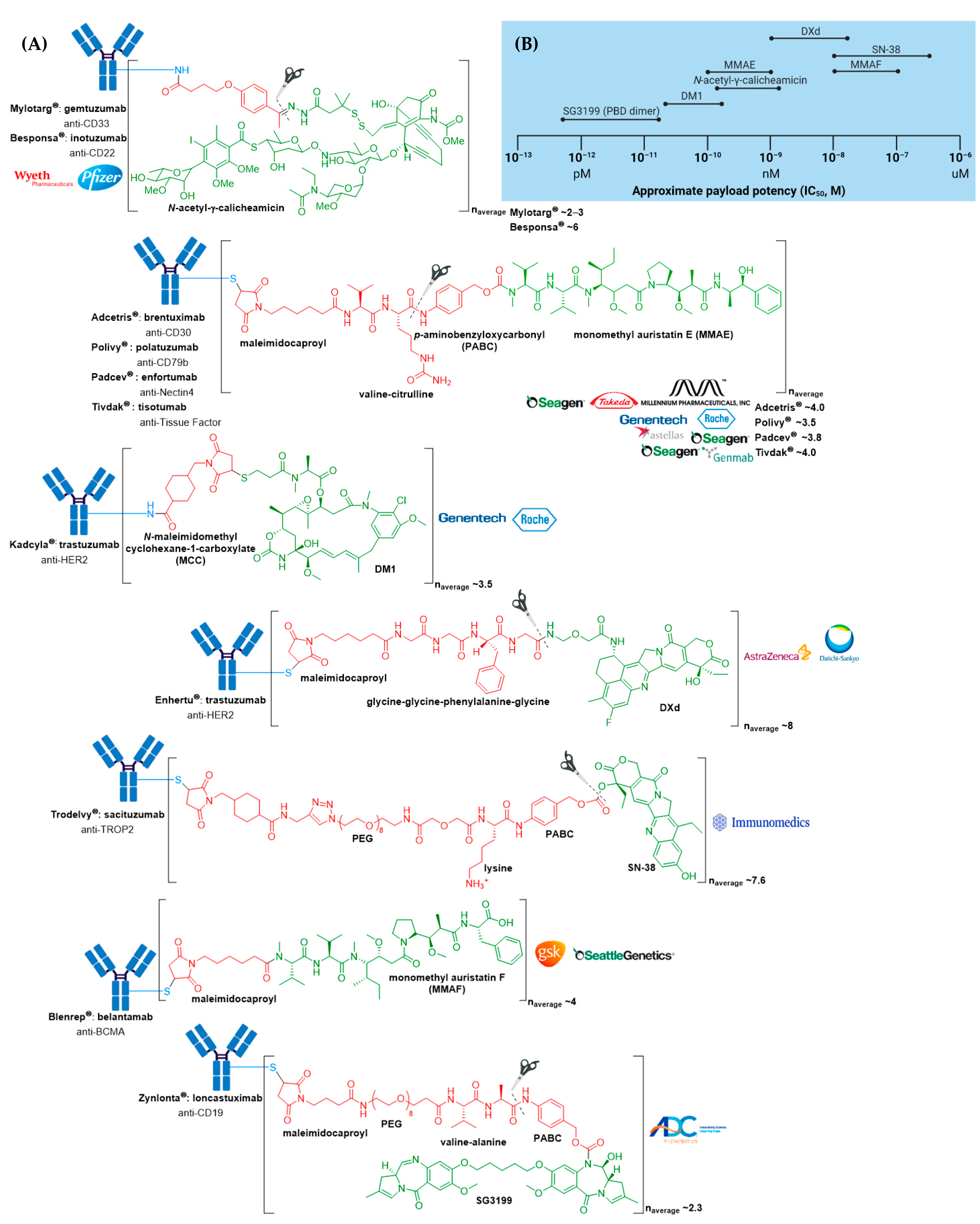
3. FDA Approved ADCs
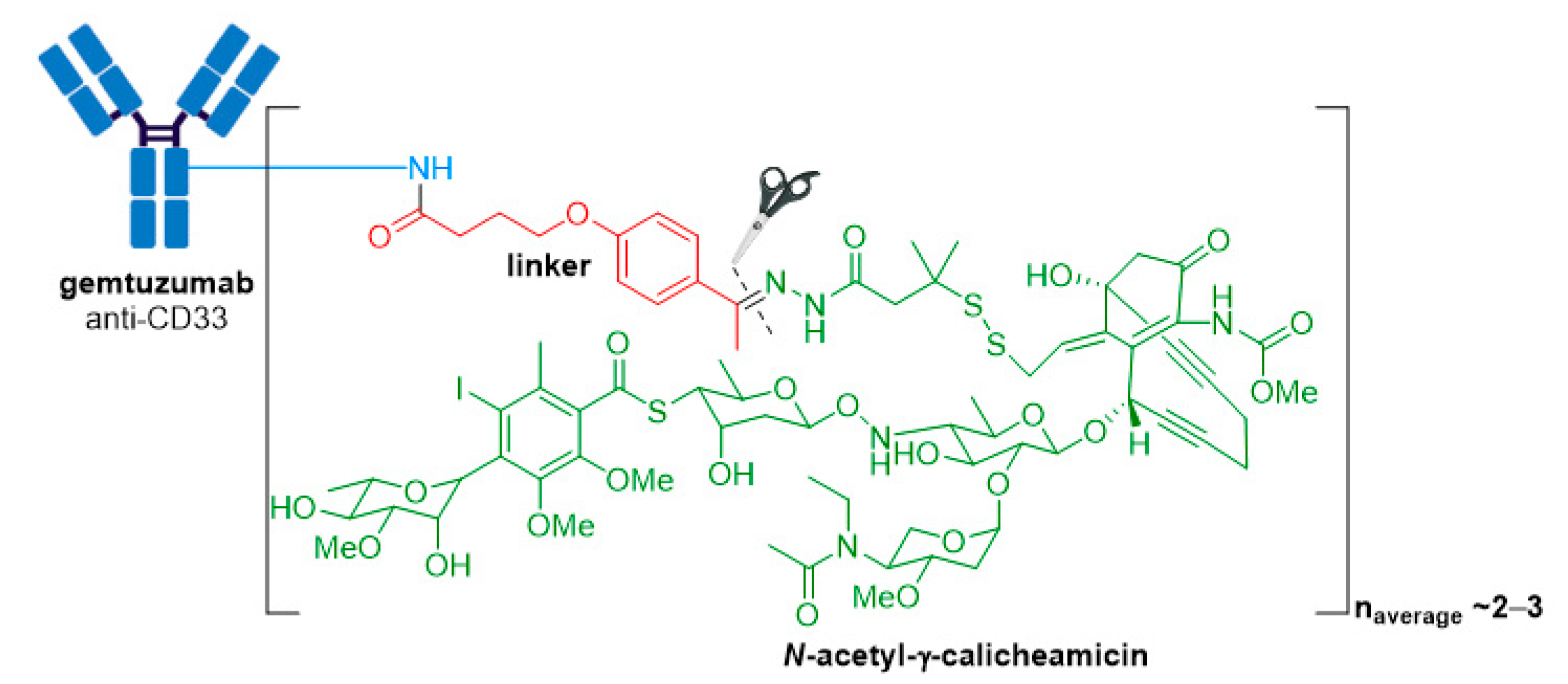
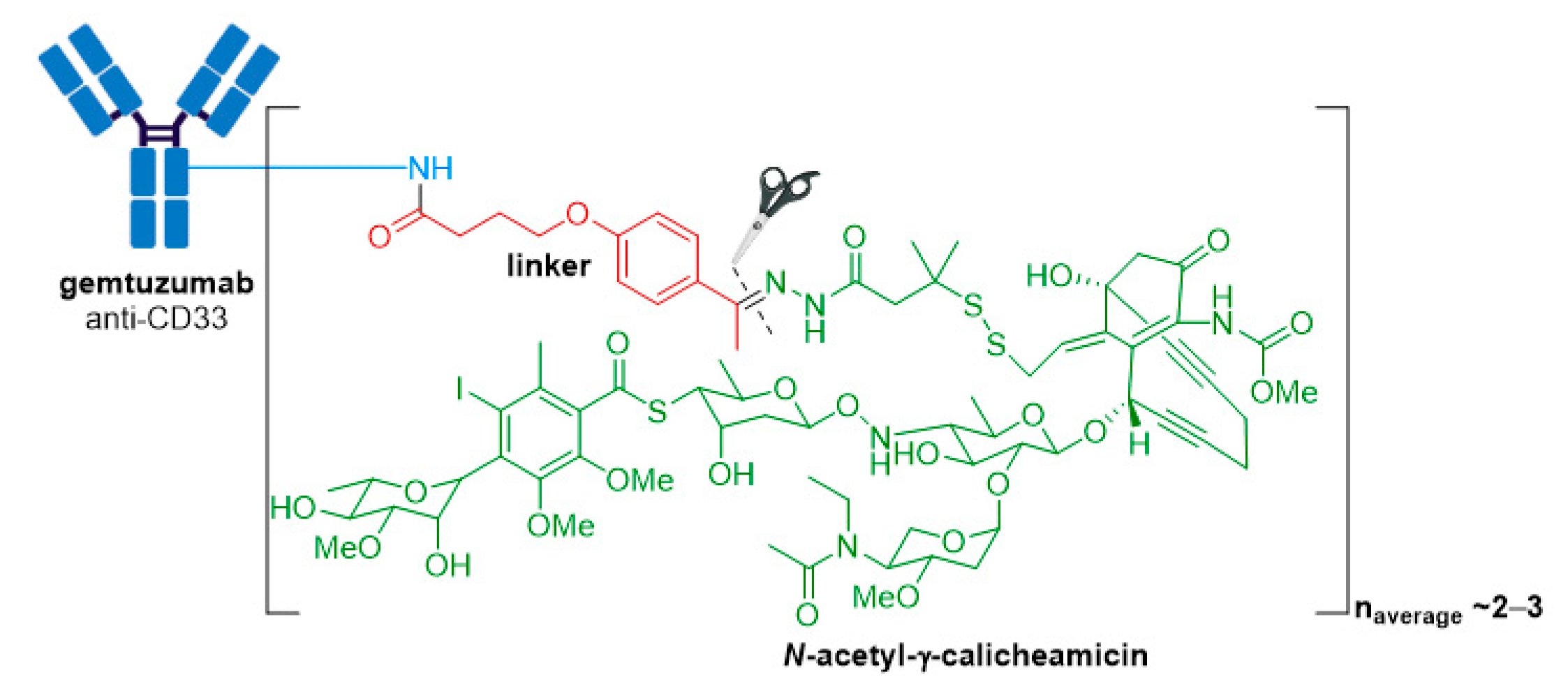
3.1. Mylotarg®
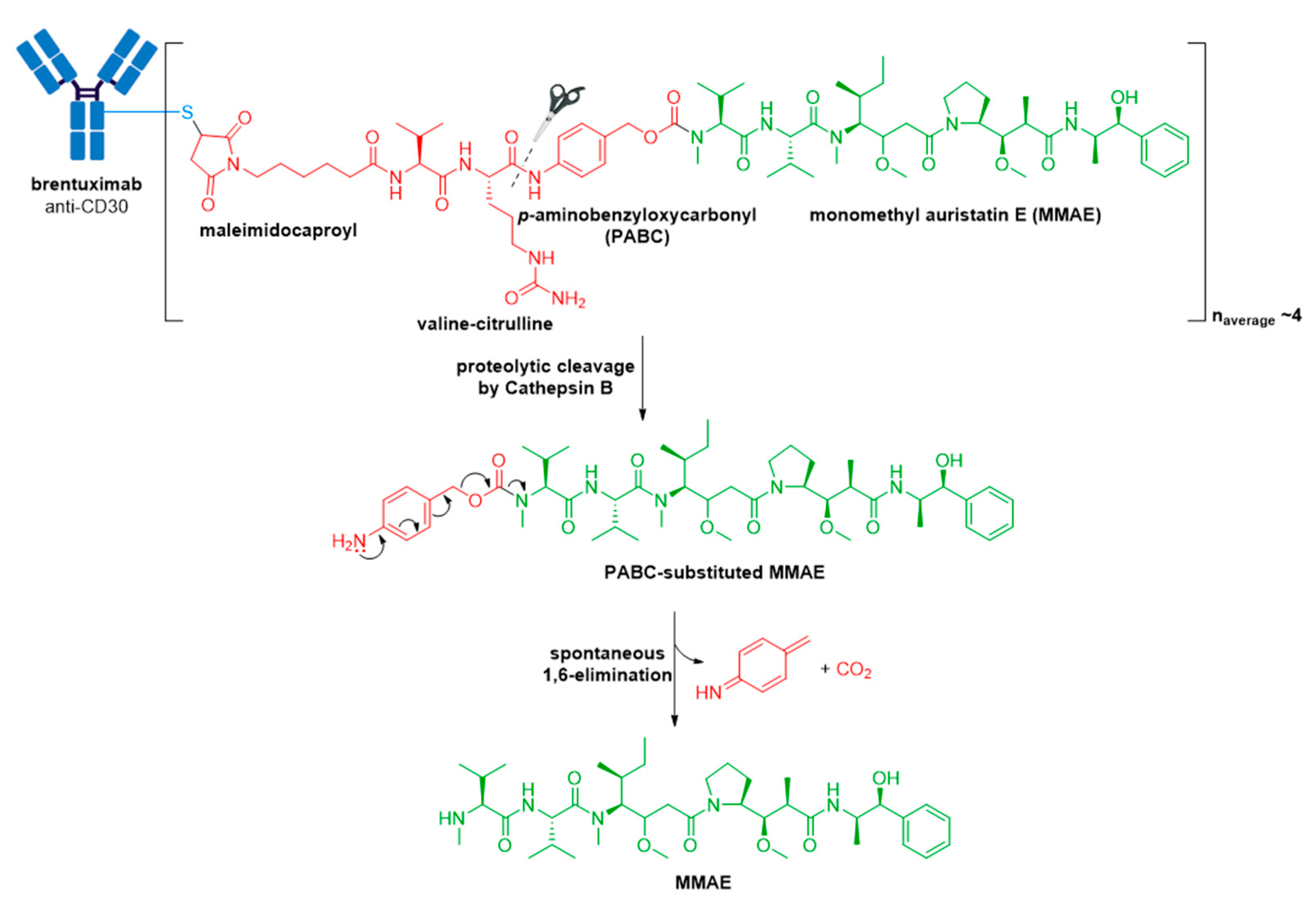
3.2. Adcetris®
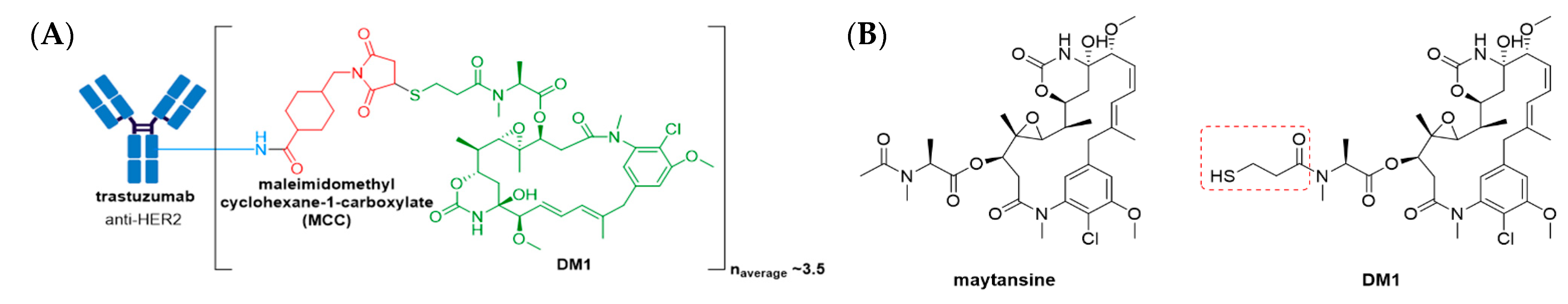
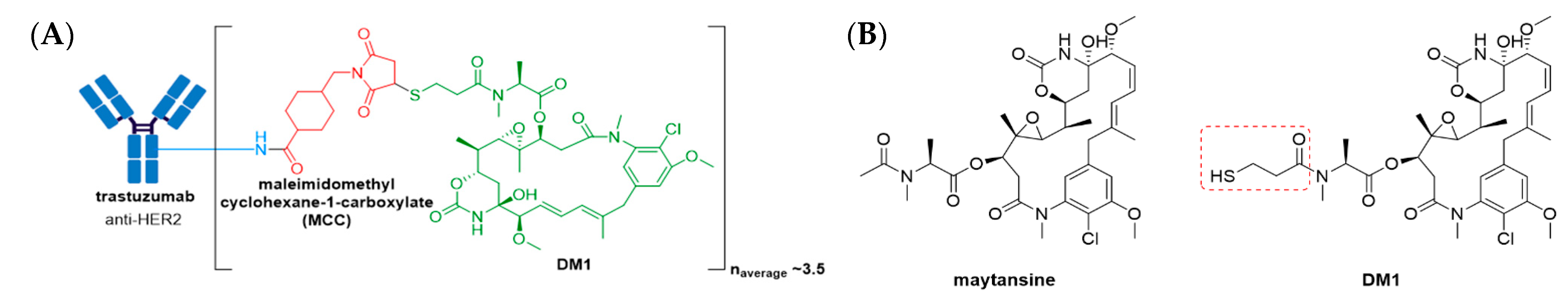
3.3. Kadcyla®
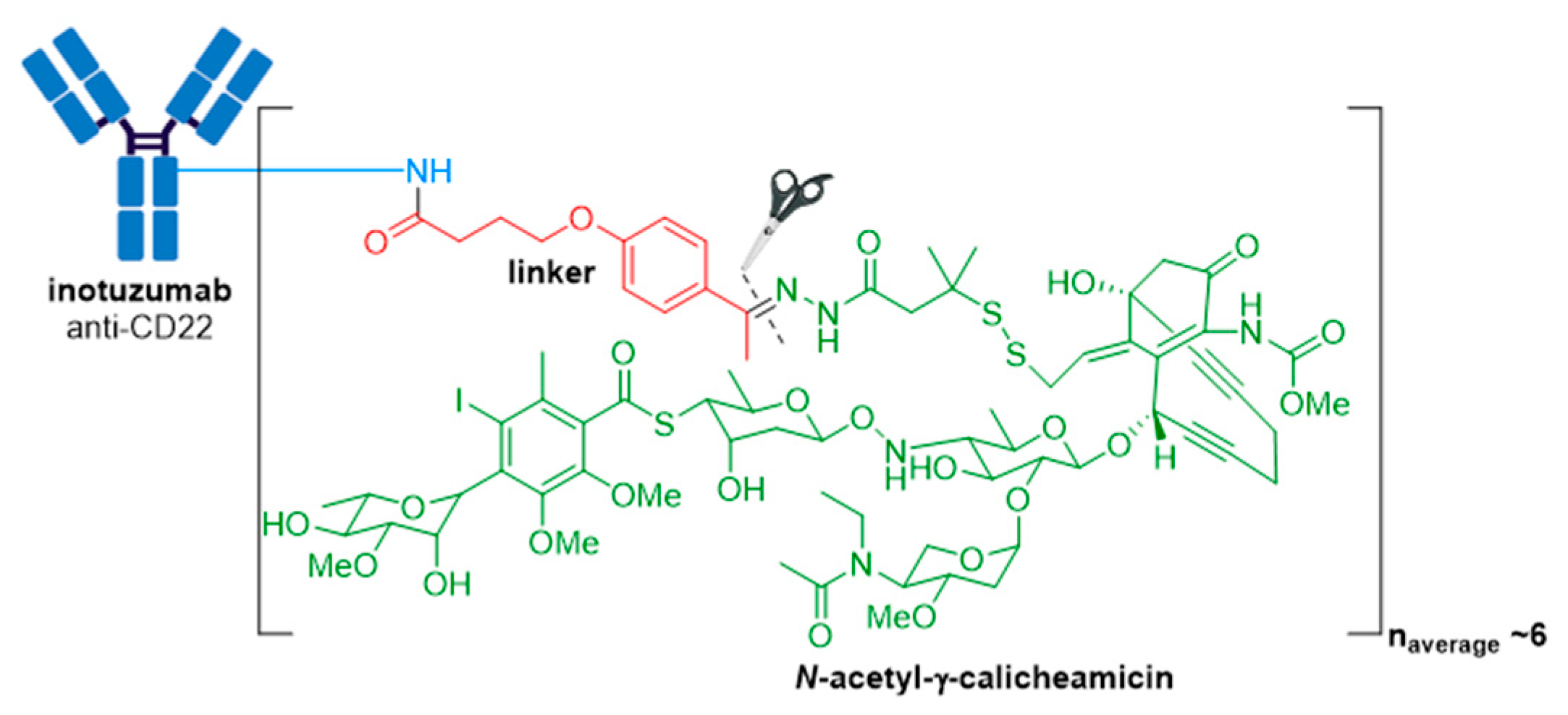
3.4. Besponsa®
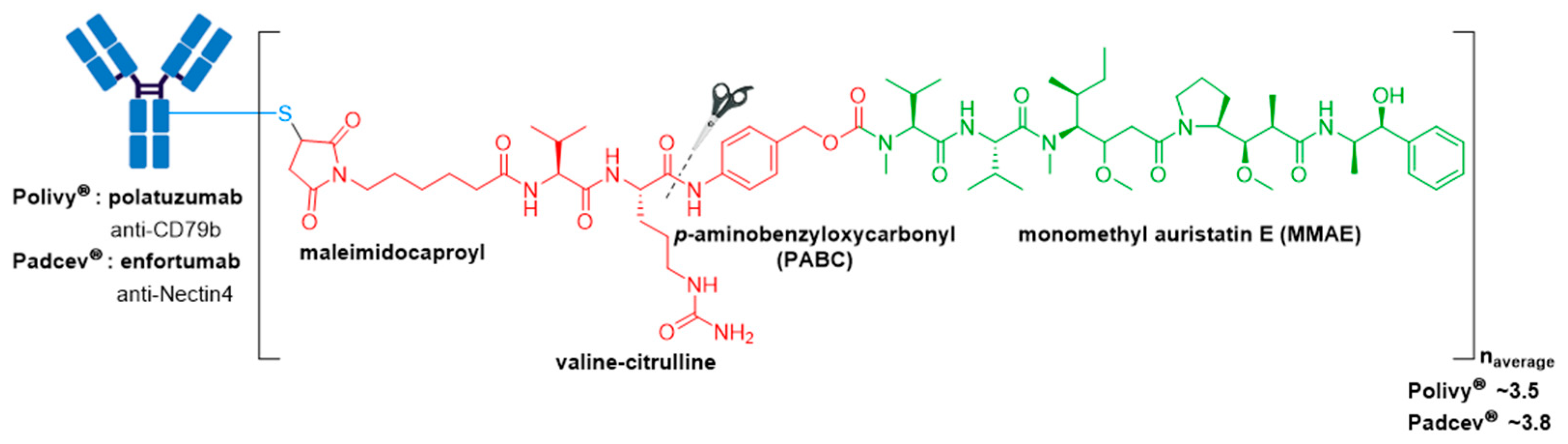
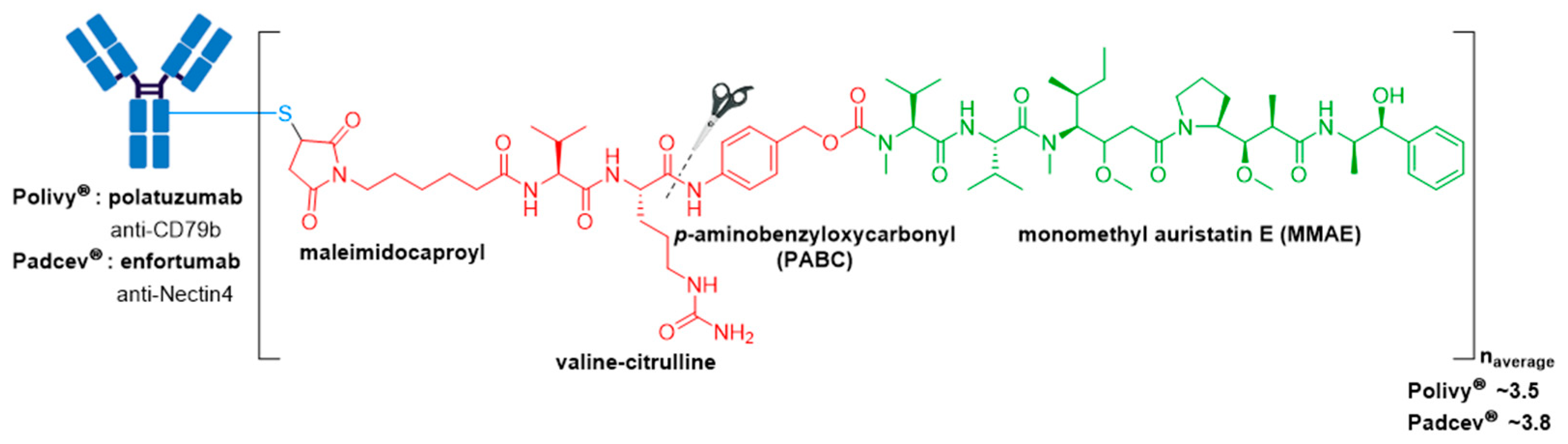
3.5. Polivy® and Padcev®
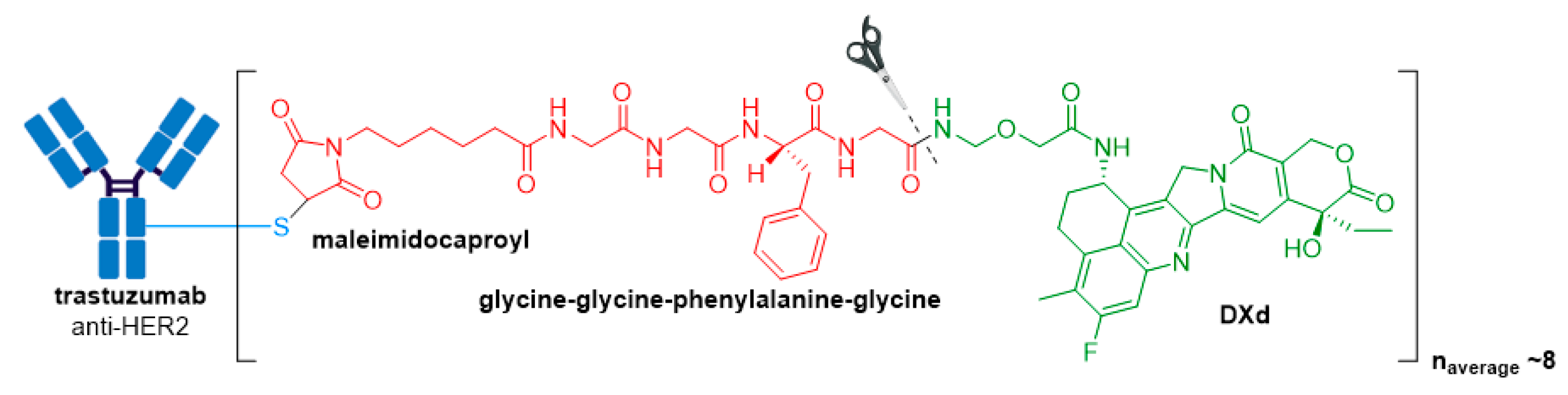
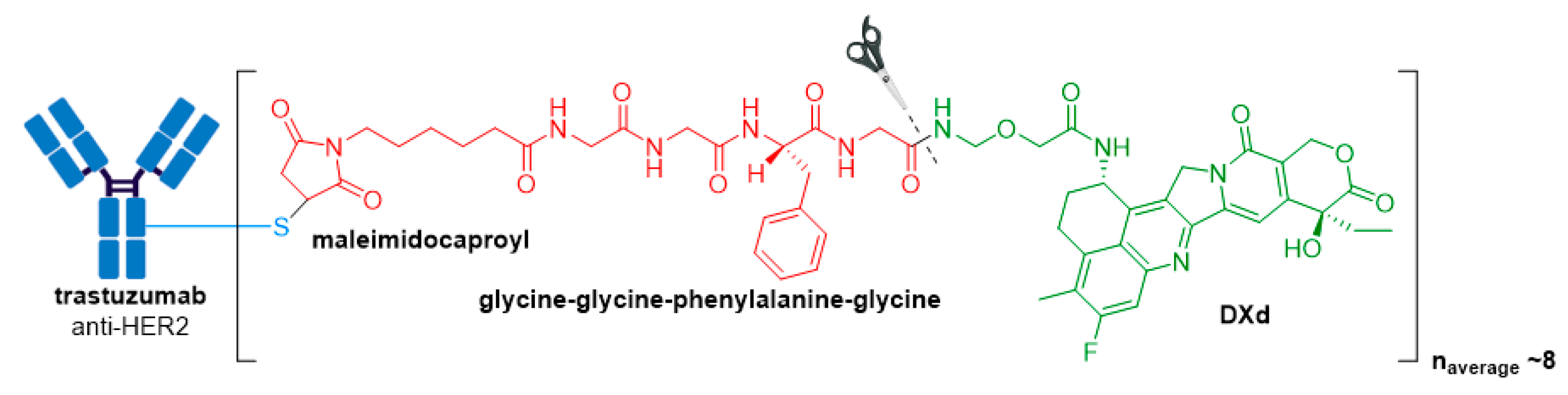
3.6. Enhertu®
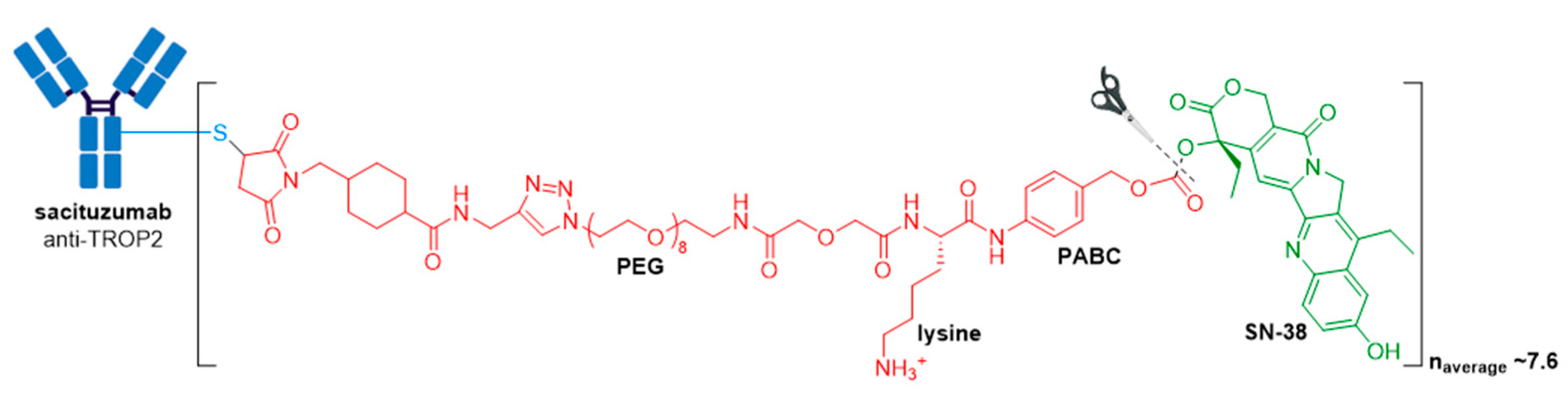
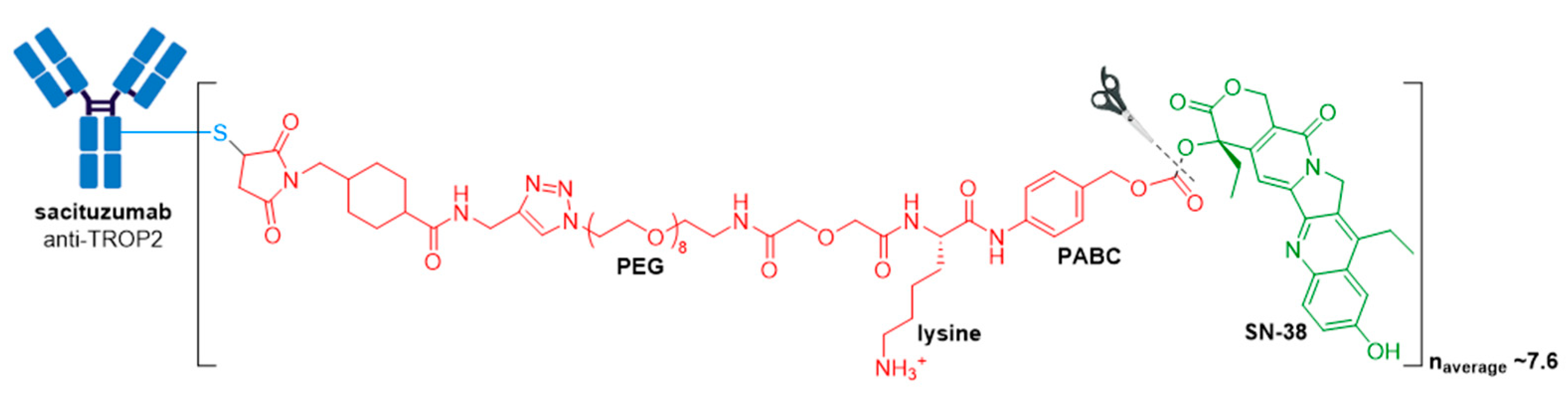
3.7. Trodelvy®
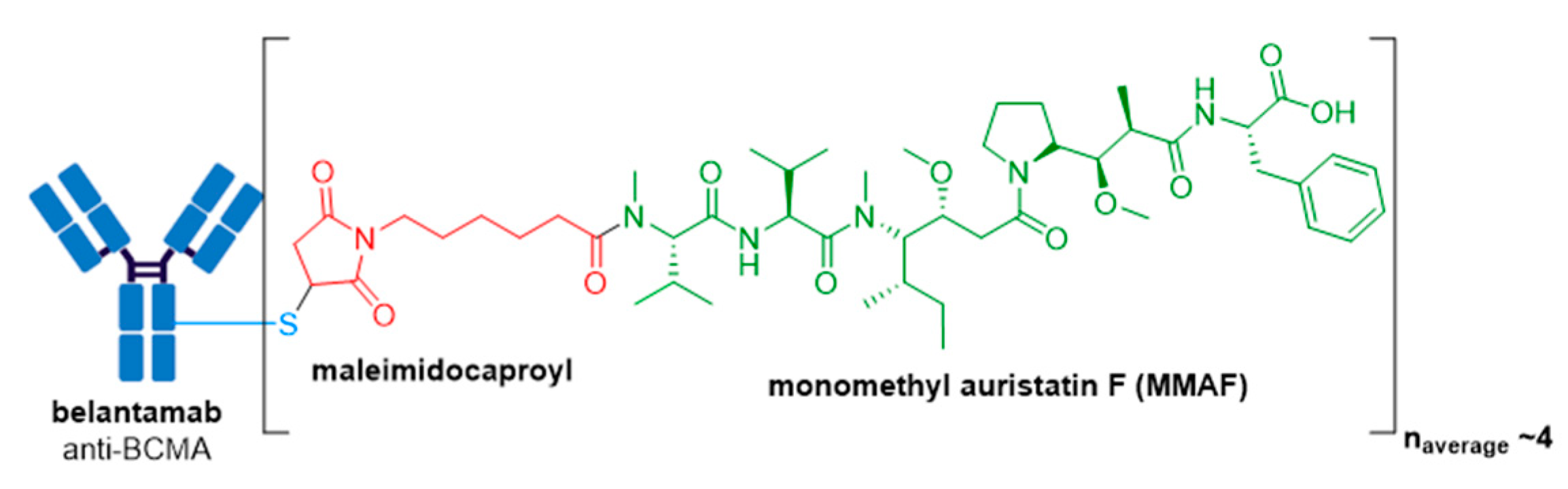
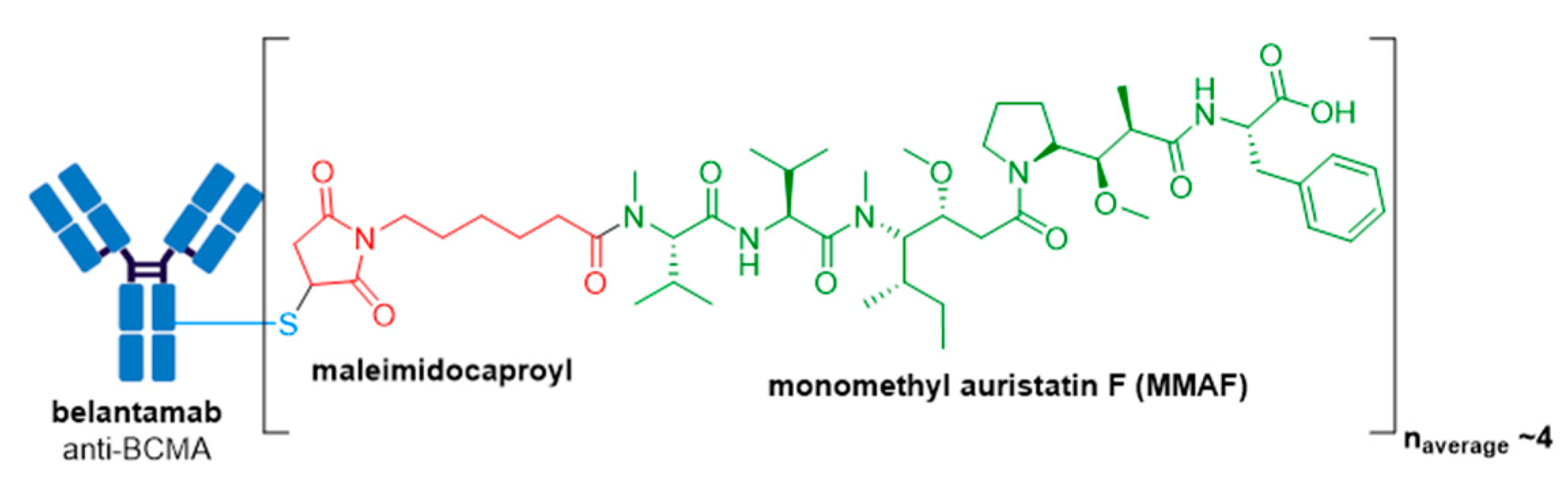
3.8. Blenrep®
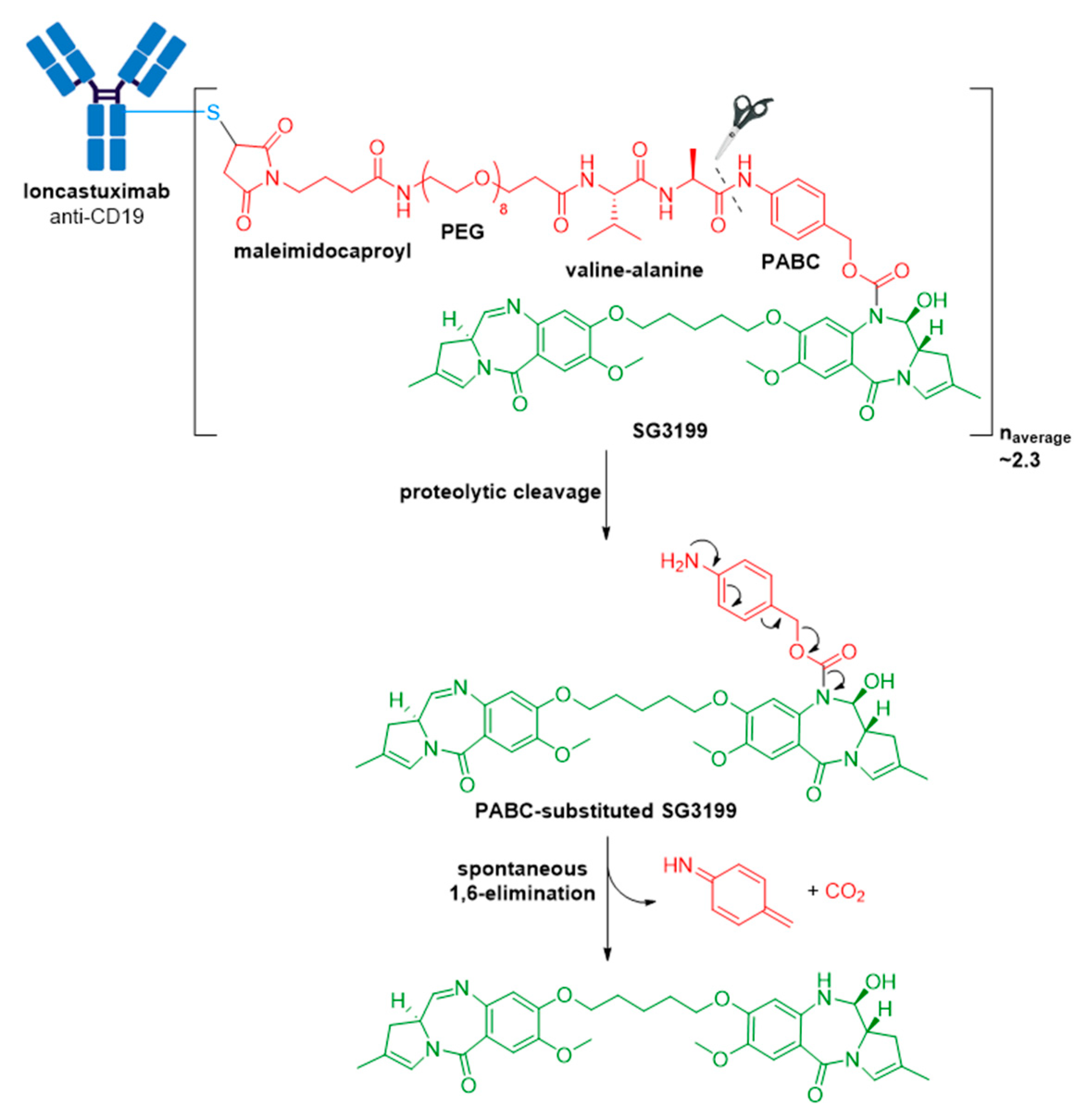
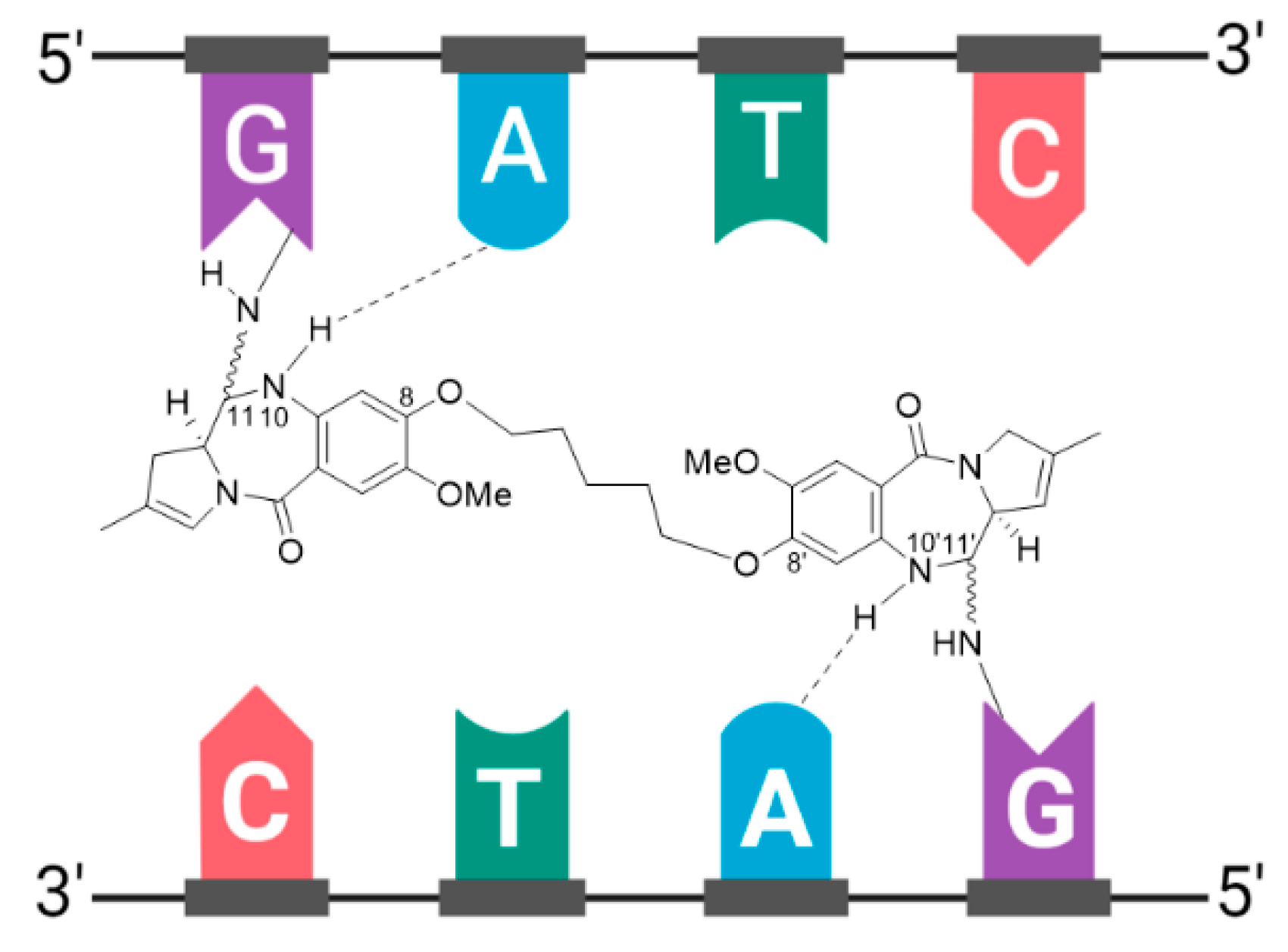
3.9. Zynlonta®
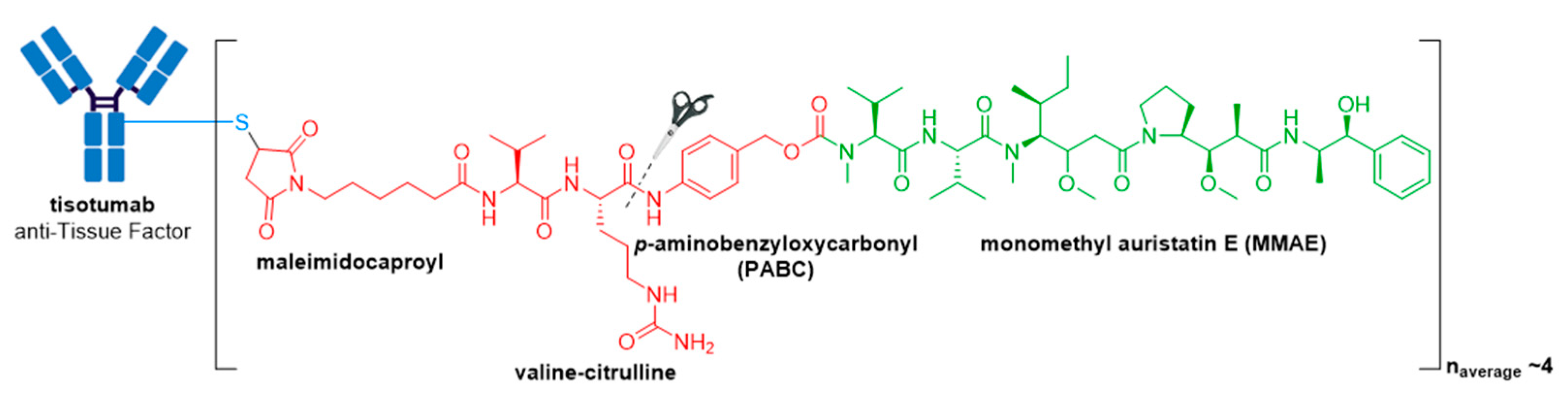
3.10. Tivdak®
4. Future Outlook and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix B
References
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Ford, C.H.; Newman, E.C.; Johnson, J.R.; Woodhouse, C.S.; Reeder, A.T.; Rowland, G.F.; Simmonds, R.G. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br. J. Cancer 1983, 47, 35–42. [Google Scholar] [CrossRef]
- Tolcher, A.W. The evolution of antibody-drug conjugates: A positive inflexion point. Am. Soc. Clin. Oncol. Educ. Book 2020, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Sievers, E.L.; Senter, P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef]
- Pazo, C.D.; Nawaz, K.; Webster, R.M. The oncology market for antibody–drug conjugates. Nat. Rev. Drug Discov. 2021, 20, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Kostova, V.; Désos, P.; Starck, J.-B.; Kotschy, A. The chemistry behind ADCs. Pharmaceuticals 2021, 14, 442. [Google Scholar] [CrossRef]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins 2011, 3, 848–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, R.; Pastan, I. Immunogenicity of immunotoxins containing pseudomonas exotoxin A: Causes, consequences, and mitigation. Front. Immunol. 2020, 11, 1261. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Bruins, W.S.C.; Zweegman, S.; Mutis, T.; Van De Donk, N.W.C.J. Targeted therapy with immunoconjugates for multiple myeloma. Front. Immunol. 2020, 11, 1155. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. LUMOXITI (Moxetumomab Pasudotox): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761104s000lbl.pdf (accessed on 20 September 2021).
- AstraZeneca. US FDA Approves Lumoxiti (Moxetumomab Pasudotox-Tdfk) for Certain Patients with Relapsed or Refractory Hairy Cell Leukaemia. Available online: https://www.astrazeneca.com/media-centre/press-releases/2018/us-fda-approves-lumoxiti-moxetumomab-pasudotox-tdfk-for-certain-patients-with-relapsed-or-refractory-hairy-cell-leukaemia.html (accessed on 21 September 2021).
- U.S. Food and Drug Administration. ONTAK (Denileukin Difitox): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/103767s5144lbl.pdf (accessed on 20 September 2021).
- U.S. Food and Drug Administration. ELZONRIS (Tagraxofusp-Erzs): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761116s000lbl.pdf (accessed on 20 September 2021).
- Pysz, I.; Jackson, P.J.M.; Thurston, D.E. Introduction to Antibody–Drug Conjugates (ADCs); The Royal Society of Chemistry: London, UK, 2019; Chapter 1; pp. 1–30. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
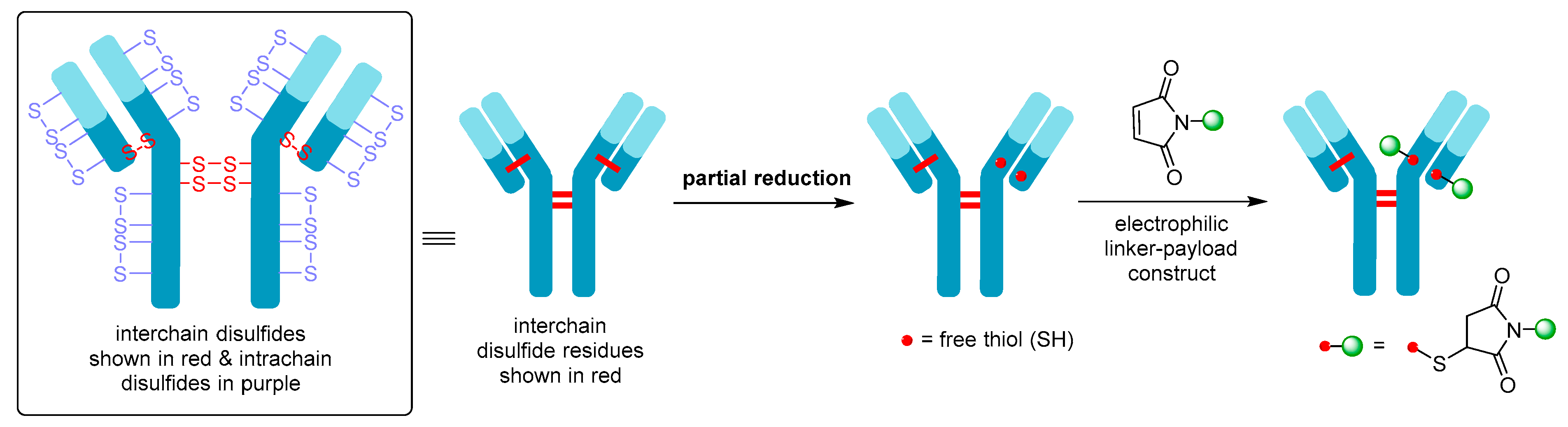
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody–drug conjugates. Chem. Soc. Rev. 2020, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–drug conjugates: A comprehensive review. Mol. Cancer Res. 2019, 18, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013, 5, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.; Nickerson, D.P.; Odorizzi, G. Molecular mechanisms of late endosome morphology, identity and sorting. Curr. Opin. Cell Biol. 2006, 18, 422–428. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. MYLOTARG (Gemtuzumab Ozogamicin): US Prescribing Information 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761060s004lbl.pdf (accessed on 11 August 2021).
- Naito, K.; Takeshita, A.; Shigeno, K.; Nakamura, S.; Fujisawa, S.; Shinjo, K.; Yoshida, H.; Ohnishi, K.; Mori, M.; Terakawa, S.; et al. Calicheamicin-conjugated humanized anti-CD33 monoclonal antibody (gemtuzumab zogamicin, CMA-676) shows cytocidal effect on CD33-positive leukemia cell lines, but is inactive on P-glycoprotein-expressing sublines. Leukemia 2000, 14, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. FDA Approves Mylotarg for Treatment of Acute Myeloid Leukemia. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-mylotarg-treatment-acute-myeloid-leukemia (accessed on 1 September 2021).
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: Results of the randomized phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef]
- Taksin, A.-L.; Legrand, O.; Raffoux, E.; De Revel, T.; Thomas, X.; Contentin, N.; Bouabdallah, R.; Pautas, C.; Turlure, P.; Reman, O.; et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: A prospective study of the alfa group. Leukemia 2006, 21, 66–71. [Google Scholar] [CrossRef]
- Renneville, A.; Abdelali, B.R.; Chevret, S.; Nibourel, O.; Cheok, M.; Pautas, C.; Duléry, R.; Boyer, T.; Cayuela, J.-M.; Hayette, S.; et al. Clinical impact of gene mutations and lesions detected by SNP-array karyotyping in acute myeloid leukemia patients in the context of gemtuzumab ozogamicin treatment: Results of the ALFA-0701 trial. Oncotarget 2014, 5, 916–932. [Google Scholar] [CrossRef] [Green Version]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: Results from the randomized phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, P.; Basak, A.; Campbell, A.; Alabugin, I.V. Photochemical activation of enediyne warheads: A potential tool for targeted antitumor therapy. Mol. Pharm. 2018, 15, 768–797. [Google Scholar] [CrossRef]
- Vollmar, B.S.; Frantz, C.; Schutten, M.M.; Zhong, F.; del Rosario, G.; Go, M.A.T.; Yu, S.-F.; Leipold, D.D.; Kamath, A.V.; Ng, C.; et al. Calicheamicin antibody–drug conjugates with improved properties. Mol. Cancer Ther. 2021, 20, 1112–1120. [Google Scholar] [CrossRef]
- Hinman, L.M.; Hamann, P.R.; Wallace, R.; Menendez, A.T.; Durr, F.E.; Upeslacis, J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: A novel and potent family of antitumor antibiotics. Cancer Res. 1993, 53, 3336–3342. [Google Scholar]
- Kim, J.H.; Hong, H.J. Humanization by CDR grafting and specificity-determining residue grafting. Antib. Eng. 2012, 907, 237–245. [Google Scholar] [CrossRef]
- Williams, D.G.; Matthews, D.J.; Jones, T. Humanising Antibodies by CDR Grafting; Springer: Berlin/Heidelberg, Germany, 2010; pp. 319–339. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of antibody drug conjugates (ADCs). Oncoimmunology 2017, 7, e1395127. [Google Scholar] [CrossRef] [PubMed]
- Lazar, G.A.; Dang, W.; Karki, S.; Vafa, O.; Peng, J.S.; Hyun, L.; Chan, C.; Chung, H.S.; Eivazi, A.; Yoder, S.C.; et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 2006, 103, 4005–4010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.R.; Woof, J.M. Human antibody effector function. Adv. Immunol. 1992, 51, 1–84. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. ADCETRIS (Brentuximab Vedotin): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125388s099lbl.pdf (accessed on 20 September 2021).
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; Deblanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, A.; Picardi, M.; et al. Brentuximab vedotin with chemotherapy for Stage III or IV Hodgkin’s lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.I.; Chen, A.; Stiff, P.; Gianni, A.M.; Carella, A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, A.; Bartlett, N.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.; Sussman, D.; Prota, A.E. Structural basis of microtubule destabilization by potent auristatin anti-mitotics. PLoS ONE 2016, 11, e0160890. [Google Scholar] [CrossRef] [Green Version]
- Oflazoglu, E.; Stone, I.J.; Gordon, K.A.; Grewal, I.S.; van Rooijen, N.; Law, C.-L.; Gerber, H.-P. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood 2007, 110, 4370–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2007, 41, 98–107. [Google Scholar] [CrossRef]
- Hughes, B. Antibody–drug conjugates for cancer: Poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [Google Scholar] [CrossRef]
- Oflazoglu, E.; Kissler, K.M.; Sievers, E.; Grewal, I.; Gerber, H.-P. Combination of the anti-CD30-auristatin-E antibody-drug conjugate (SGN-35) with chemotherapy improves antitumour activity in Hodgkin lymphoma. Br. J. Haematol. 2008, 142, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; White, J.; Christie, R.J.; Dimasi, N.; Gao, C. Antibody-drug conjugates. Annu. Rep. Med Chem. 2017, 50, 441–480. [Google Scholar] [CrossRef]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2—Positive breast cancer. Clin. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Chari, R.V.J. Ado-trastuzumab emtansine (T-DM1): An antibody–drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. KADCYLA (Ado-Trastuzumab Emtansine): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125427s108lbl.pdf (accessed on 20 September 2021).
- Criscitiello, C.; Morganti, S.; Curigliano, G. Antibody–drug conjugates in solid tumors: A look into novel targets. J. Hematol. Oncol. 2021, 14, 20. [Google Scholar] [CrossRef]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [Green Version]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. BESPONSA (Inotuzumab Ozogamicin): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf (accessed on 20 September 2021).
- Di Joseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-targeted chemotherapy with CMC-544: A CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Food and Drug Administration. POLIVY (Polatuzumab Vedotin-Piiq). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 20 September 2021).
- U.S. Food and Drug Administration. PADCEV (Enfortumab Vedotin-Ejfv): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761137s006s008lbl.pdf (accessed on 20 September 2021).
- Palanca-Wessels, M.M.C.; Salles, G.A.; Czuczman, M.S.; Assouline, S.E.; Flinn, I.W.; Sehn, L.H.; Patel, M.; Sangha, R.; Tilly, H.; Advani, R.; et al. Final results of a phase i study of the anti-CD79b antibody-drug conjugate DCDS4501A in relapsed or refractory (R/R) B-cell non-hodgkin lymphoma (NHL). Blood 2013, 122, 4400. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab vedotin in relapsed or refractory diffuse large B-cell lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; Rossana, N.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab vedotin antibody–drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef]
- Yu, E.Y.; Petrylak, D.P.; O’Donnell, P.H.; Lee, J.-L.; van der Heijden, M.S.; Loriot, Y.; Stein, M.N.; Necchi, A.; Kojima, T.; Harrison, M.R.; et al. Enfortumab vedotin after PD-1 or PD-L1 inhibitors in cisplatin-ineligible patients with advanced urothelial carcinoma (EV-201): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 872–882. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J. Clin. Oncol. 2019, 37, 2592–2600. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. ENHERTU (Fam-Trastuzumab Deruxtecan-Nxki): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761139s011lbl.pdf (accessed on 20 September 2021).
- Shitara, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.-C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Sankyo, D. Daiichi Sankyo and AstraZeneca Enter New Global Development and Commercialization Collaboration for Daiichi Sankyo’s ADC DS-1062. Available online: https://www.daiichisankyo.com/media/press_release/detail/index_3126.html (accessed on 31 August 2021).
- AstraZeneca. AstraZeneca and Daiichi Sankyo Enter Collaboration to Develop and Commercialize New Antibody Drug Conjugate. Available online: https://www.astrazeneca.com/media-centre/press-releases/2020/astrazeneca-and-daiichi-sankyo-enter-collaboration-to-develop-and-commercialise-new-antibody-drug-conjugate.html (accessed on 31 August 2021).
- Businesswire. Gilead Sciences to Acquire Immunomedics. Available online: https://www.businesswire.com/news/home/20200913005051/en/Gilead-Sciences-to-Acquire-Immunomedics (accessed on 31 August 2021).
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. TRODELVY (Sacituzumab Govitecan-Hziy): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761115s009lbl.pdf (accessed on 20 September 2021).
- Gilead. Gilead Sciences Announces Fourth Quarter and Full Year 2020 Financial Results. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2021/2/gilead-sciences-announces-fourth-quarter-and-full-year-2020-financial-results (accessed on 31 August 2021).
- Tagawa, S.T.; Balar, A.V.; Petrylak, D.P.; Kalebasty, A.R.; Loriot, Y.; Fléchon, A.; Jain, R.K.; Agarwal, N.; Bupathi, M.; Barthelemy, P.; et al. TROPHY-U-01: A phase II open-label study of sacituzumab govitecan in patients with metastatic urothelial carcinoma progressing after platinum-based chemotherapy and checkpoint inhibitors. J. Clin. Oncol. 2021, 39, 2474–2485. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab govitecan: First approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Goldenberg, D.M. Humanized anti-trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: Preclinical studies in human cancer xenograft models and monkeys. Clin. Cancer Res. 2011, 17, 3157–3169. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 2015, 6, 22496–22512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GSK. FDA Approves GSK’s BLENREP (Belantamab Mafodotin-Blmf) for the Treatment of Patients with Relapsed or Refractory Multiple Myeloma. Available online: https://www.gsk.com/en-gb/media/press-releases/fda-approves-gsk-s-blenrep-belantamab-mafodotin-blmf-for-the-treatment-of-patients-with-relapsed-or-refractory-multiple-myeloma/ (accessed on 31 August 2021).
- U.S. Food and Drug Administration. BLENREP (Belantamab Mafodotin-Blmf): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf (accessed on 20 September 2021).
- Markham, A. Belantamab mafodotin: First approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2019, 21, 207–221. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.R.W.; Gliddon, L.; Fieles, W.; et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Yamane-Ohnuki, N.; Kinoshita, S.; Inoue-Urakubo, M.; Kusunoki, M.; Iida, S.; Nakano, R.; Wakitani, M.; Niwa, R.; Sakurada, M.; Uchida, K.; et al. Establishment ofFUT8 knockout Chinese hamster ovary cells: An ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol. Bioeng. 2004, 87, 614–622. [Google Scholar] [CrossRef]
- Pereira, N.A.; Chan, K.F.; Lin, P.C.; Song, Z. The “less-is-more” in therapeutic antibodies: Afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs 2018, 10, 693–711. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.-T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, S.O.; Senter, P.D. Auristatin Payloads for Antibody–Drug Conjugates (ADCs); The Royal Society of Chemistry: London, UK, 2019; Chapter 4; pp. 73–99. [Google Scholar] [CrossRef]
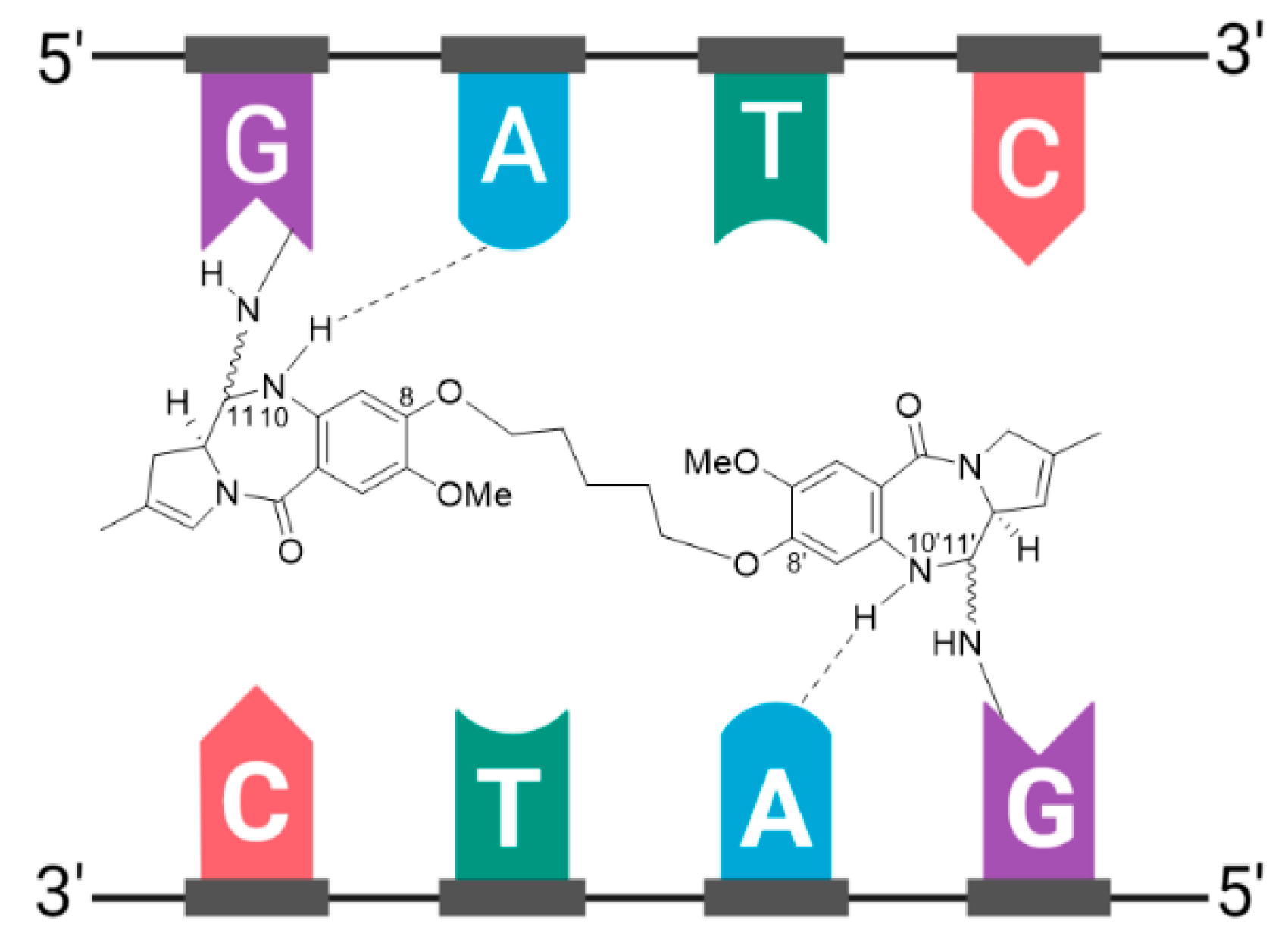
- U.S. Food and Drug Administration. ZYNLONTA (Loncastuximab Tesirine-Lpyl). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf (accessed on 20 September 2021).
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef]
- Zammarchi, F.; Corbett, S.; Adams, L.; Tyrer, P.C.; Kiakos, K.; Janghra, N.; Marafioti, T.; Britten, C.E.; Havenith, C.E.G.; Chivers, S.; et al. ADCT-402, a PBD dimer–containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 2018, 131, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. TIVDAK (Tisotumab Vedotin-Tftv): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761208s000lbl.pdf (accessed on 22 September 2021).
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef]
- De Goeij, B.E.; Satijn, D.; Freitag, C.M.; Wubbolts, R.; Bleeker, W.K.; Khasanov, A.; Zhu, T.; Chen, G.; Miao, D.; Van Berkel, P.H.; et al. High turnover of tissue factor enables efficient intracellular delivery of antibody–drug conjugates. Mol. Cancer Ther. 2015, 14, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Freedman, D.H. Hunting for new drugs with AI. Nature 2019, 576, S49–S53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, N. Tapping into the drug discovery potential of AI. Biopharma Deal. 2021. [Google Scholar] [CrossRef]
- Wolfe, J.M.; Fadzen, C.M.; Choo, Z.-N.; Holden, R.L.; Yao, M.; Hanson, G.J.; Pentelute, B.L. Machine learning to predict cell-penetrating peptides for antisense delivery. ACS Cent. Sci. 2018, 4, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Mohapatra, S.; Hartrampf, N.; Poskus, M.; Loas, A.; Gómez-Bombarelli, R.; Pentelute, B.L. Deep Learning for Prediction and Optimization of Fast-Flow Peptide Synthesis. ACS Cent. Sci. 2020, 6, 2277–2286. [Google Scholar] [CrossRef]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]
- Alyass, A.; Turcotte, M.; Meyre, D. From big data analysis to personalized medicine for all: Challenges and opportunities. BMC Med. Genom. 2015, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef]
- Wakankar, A.; Chen, Y.; Gokarn, Y.; Jacobson, F.S. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 2011, 3, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Neupane, R.; Bergquist, J. Analytical techniques for the characterization of antibody drug conjugates: Challenges and prospects. Eur. J. Mass Spectrom. 2017, 23, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J. Drug-to-antibody ratio (DAR) and drug load distribution by hydrophobic interaction chromatography and reversed phase high-performance liquid chromatography. Methods Mol. Biol. 2013, 1045, 275–283. [Google Scholar] [CrossRef]
- McPherson, M.J.; Hobson, A. Pushing the envelope: Advancement of ADCs outside of oncology. Methods Mol. Biol. 2019, 2078, 23–36. [Google Scholar] [CrossRef]
- Fernandez-Codina, A.; Nevskaya, T.; Pope, J. OP0172 brentuximab vedontin for skin involvement in refractory diffuse cutaneous systemic sclerosis, interim results of A phase IIB open-label trial. Ann. Rheum. Dis. 2021, 80, 103–104. [Google Scholar] [CrossRef]
- Pushpakom, S. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Cameron, T.; Morrison, C. 2020 biotech IPOs shatter all the records. Nat. Rev. Drug Discov. 2021, 20, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Hardison, S. Oncology dealmaking in 2020. Biopharma Deal. 2021. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC | Target | mAb | Linker | Payload/Payload Class | Payload Action | DAR | Disease Indication (Year of Approval) |
|---|---|---|---|---|---|---|---|
| Mylotarg® (gemtuzumab ozogamicin) | CD33 | IgG4 | acid cleavable | ozogamicin/calicheamicin | DNA cleavage | 2–3 | CD33+ R/R AML (2000) a |
| Adcetris® (brentuximab vedotin) | CD30 | IgG1 | enzyme cleavable | MMAE/auristatin | microtubule inhibitor | 4 | R/R sALCL or cHL (2011)R/R pcALCL or CD30+ MF (2017); cHL, sALCL or CD30+ PTCL (2018) b |
| Kadcyla® (ado-trastuzumab emtansine) | HER2 | IgG1 | non-cleavable | DM1/maytansinoid | microtubule inhibitor | 3.5 | HER2+ metastatic breast cancer previously treated with trastuzumab & a taxane (2013); HER2+ early breast cancer after neoadjuvant taxane & trastuzumab-based treatment (2019) |
| Besponsa® (inotuzumab ozogamicin) | CD22 | IgG4 | acid cleavable | ozogamicin/calicheamicin | DNA cleavage | 6 | R/R B-ALL (2017) |
| Polivy® (polatuzumab vedotin-piiq) | CD79b | IgG1 | enzyme cleavable | MMAE/auristatin | microtubule inhibitor | 3.5 | R/R DLBCL (2019) c,d |
| Padcev® (enfortumab vedotin-ejfv) | Nectin4 | IgG1 | enzyme cleavable | MMAE/auristatin | microtubule inhibitor | 3.8 | Locally advanced or metastatic urothelial cancer after a PD-1 or PD-L1 inhibitor and a Pt-containing chemotherapy (2019) or are ineligible for cisplatin-containing chemotherapy and previously received 1 or more lines of therapy (2021) d |
| Enhertu® (fam-trastuzumab deruxtecan-nxki) | HER2 | IgG1 | enzyme cleavable | DXd/camptothecin | TOP1 inhibitor | 8 | Unresectable or metastatic HER2+ breast cancer after 2 or more anti-HER2 regimens (2019) d; locally advanced or metastatic HER2+ gastric or gastroesophageal junction adenocarcinoma after a trastuzumab-based regimen (2021) |
| Trodelvy® (sacituzumab govitecan-hziy) | TROP2 | IgG1 | acid cleavable | SN-38/camptothecin | TOP1 inhibitor | 7.6 | Locally advanced or metastatic TNBC after at least two prior therapies (2020); locally advanced or metastatic urothelial cancer after a Pt-containing chemotherapy and a PD-1 or PD-L1 inhibitor (2021) d |
| Blenrep® (belantamab mafodotin-blmf) | BCMA | IgG1 | non-cleavable | MMAF/auristatin | microtubule inhibitor | 4 | R/R multiple myeloma after at least 4 prior therapies including an anti-CD38 mAb, a proteasome inhibitor, and an immunomodulatory agent (2020) d |
| Zynlonta® (loncastuximab tesirine-lpyl) | CD19 | IgG1 | enzyme cleavable | SG3199/PBD dimer | DNA cleavage | 2.3 | R/R large B-cell lymphoma after 2 or more lines of systemic therapy, including DLBCL not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma (2021) d |
| Tivdak® (tisotumab vedotin-tftv) | Tissue Factor | IgG1 | enzyme cleavable | MMAE/auristatin | microtubule inhibitor | 4 | Recurrent or metastatic cervical cancer with disease progression on or after chemotherapy (2021) d |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. https://doi.org/10.3390/molecules26195847
Tong JTW, Harris PWR, Brimble MA, Kavianinia I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules. 2021; 26(19):5847. https://doi.org/10.3390/molecules26195847
Chicago/Turabian StyleTong, Juliana T. W., Paul W. R. Harris, Margaret A. Brimble, and Iman Kavianinia. 2021. "An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy" Molecules 26, no. 19: 5847. https://doi.org/10.3390/molecules26195847
APA StyleTong, J. T. W., Harris, P. W. R., Brimble, M. A., & Kavianinia, I. (2021). An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules, 26(19), 5847. https://doi.org/10.3390/molecules26195847