Experimental
General
Melting points were determined on a hot stage instrument and are uncorrected. Infrared spectra were recorded either as KBr pellets or neat on a Perkin Elmer System 2000 FTIR. 1H-NMR spectra were recorded on a Bruker AMX300 spectrometer at 300MHz and chemical shifts are expressed in ppm using TMS as internal standard. 13C-NMR spectra were recorded on a Bruker AMX300 spectrometer at 75.4MHz and chemical shifts are expressed in ppm using chloroform as internal standard. Mass spectra were recorded on a Hewlett Packard 5898B spectrometer. Elemental analysis was performed at the Central Equipment Laboratory of the University of Northern British Columbia.
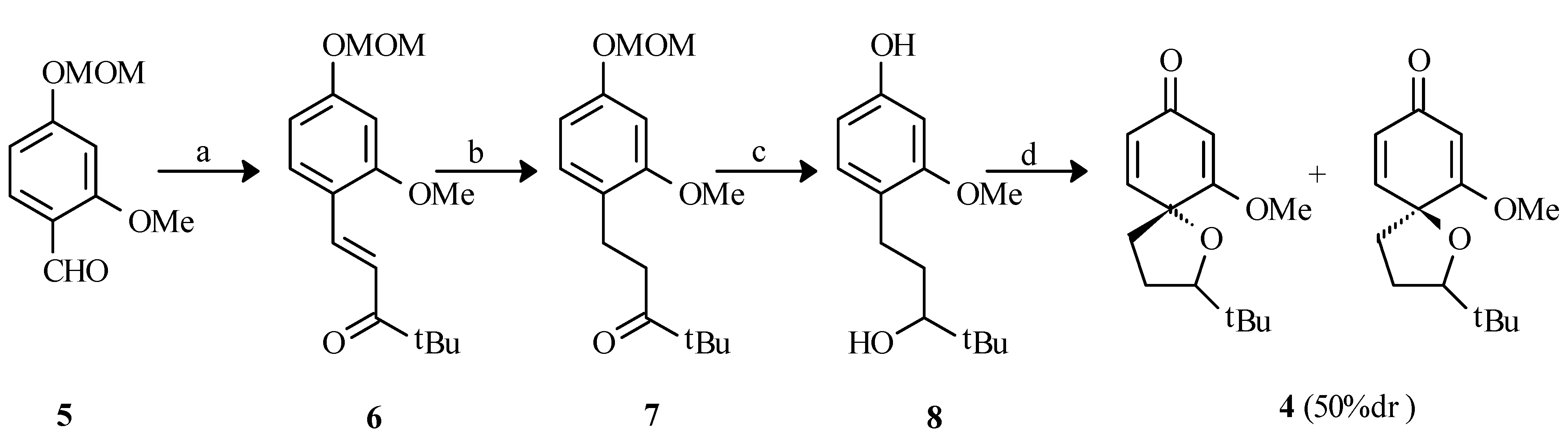
1-(2-Methoxy-4-O-methoxymethylphenyl)-4,4-dimethyl-1-penten-3-one (6).
To a solution of 2-methoxy-4-O-methoxymethylbenzaldehyde (5, 848 mg, 4.3 mmol) in 50% ethanol/tetrahydrofuran (80 mL) was added pinacolone (964 mg, 9.6 mmol) and sodium hydroxide (376 mg, 9.4 mmol). The resulting mixture was refluxed for 24 hours. Water (50 mL) was added, the solution was concentrated in vacuo to about 50% of its original volume and the aqueous residue was extracted with dichloromethane (3 x 40 mL). The organic fractions were combined, washed with brine (30 mL), dried (anhydrous MgSO4) and the solvent was evaporated in vacuo to give a yellow oil. Chromatography on silica gel (15% EtOAc/hexanes) afforded an oil (860 mg, 72%). IR (neat): 1681 (CO); 1H-NMR (CDCl3) δ: 1.22 (s, 9H, t-butyl), 3.50 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 5.25 (s, 2H, OCH2O), 6.60 (d, 1H, J=2.2Hz, Ar-H3), 6.65 (dd, 1H, J=2.2, 8.6Hz, Ar-H5), 7.13 (d, 1H, J=15.7Hz H1), 7.49 (d, 1H, J=8.6Hz, Ar-H6), 7.93 (d, 1H, J=15.7Hz, H2); 13C-NMR (CDCl3) δ: 26.7 (C5), 43.3 (C4), 55.7 (OCH3), 56.4 (OCH3), 94.5 (OCH2O), 100.2 (Ar-C3), 107.9 (Ar-C5), 118.2 (C2), 119.7 (Ar-C1), 130.5 (Ar-C6), 138.3 (C1), 160.3 (Ar-C2 and Ar-C4), 205.0 (C3); MS m/e (relative %):278 [M+] (2), 221 (100), 191 (16), 189 (16), 176 (6); Anal. Calc’d for C16H22O4: C 69.04, H 7.97; found C 69.14, H 7.91.
1-(2-Methoxy-4-O-methoxymethylphenyl)-4,4-dimethyl-3-pentanone (7)
To a solution of ketone 6 (860 mg, 3.1 mmol) in ethyl acetate (75 mL) was added Pd/C (279 mg) and the resulting mixture was stirred under a positive pressure of hydrogen for 6 hours. The suspension was filtered through Celite® and the solvent was evaporated in vacuo to give a yellowish oil. Chromatography on silica gel (20% EtOAc/hexanes) afforded a colorless oil (812 mg, 94%); IR (neat) cm-1: 1702 (CO); 1H-NMR (CDCl3) δ: 1.12 (s, 9H, t-butyl), 2.76 (m, 4H, H1 and H2), 3.48 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 5.16 (s, 2H, OCH2O), 6.56 (m, 2H, Ar-H3 and Ar-H5), 7.03 (d, 1H, J=9.1, Ar-H6); 13C-NMR (CDCl3) δ: 25.1 (C1), 25.5 (C5), 37.1 (C2), 44.3 (C4), 55.5 (OCH3), 56.2 (OCH3), 94.9 (OCH2O), 100.3 (Ar-C3), 107.2 (Ar-C5), 123.6 (Ar-C1), 130.5 (Ar-C6), 158.5 (Ar-C2 and Ar-C4), 216.0 (C3); MS m/e (relative %): 280 [M+] (51), 223 (20), 182 (12), 181 (100), 151 (52); Anal. Calc’d for C16H24O4: C 68.55, H 8.63; found C 68.43, H 8.54.
o1-(4-hydroxy-2-methoxyphenyl)-4,4-dimethyl-3-pentanol (8)
To a cold (0°C) solution of ketone 7 (810 mg, 2.9 mmol) in ethanol (50 mL) was added sodium borohydride (139 mg, 3.7 mmol). The resulting mixture was stirred at 0°C for 30 minutes then at room temperature for 2 hours. HCl (10%, 50 mL) was added and the solution was stirred at room temperature for 20 hours. The solution was concentrated in vacuo to about half its original volume and the residue was extracted with dichloromethane (3 x 40 mL), dried (MgSO4) and evaporated in vacuo to give a yellowish oil. Chromatography on silica gel (25% EtOAc/hexanes) afforded a clear oil (564 mg, 82%). IR (neat) cm-1: 3382 (OH); 1H-NMR (CDCl3) δ: 0.87 (s, 9H, t-butyl), 1.62 (m, 2H, H2), 2.69 (m, 2H, H1), 3.78 (s, 3H, OCH3), 6.35 (dd, 1H, J=2.4, 8.0Hz, Ar-H5), 6.41 (d, 1H, J=2.4Hz, Ar-H3), 6.99 (d, 1H, J=8.0Hz, Ar-H6); 13C-NMR (CDCl3) δ: 26.0 (C5), 26.6 (C2), 32.2 (C1), 34.9 (C4), 55.5 (OCH3), 79.6 (C3), 99.3 (Ar-C3), 107.3 (Ar-C5), 121.9 (Ar-C1), 130.6 (Ar-C6), 155.7 (Ar-C4), 158.3 (Ar-C2); MS m/e (relative %): 238 [M+] (8), 236 (29), 179 (12), 137 (100), 107 (12), 77 (9); Anal. Calc’d for C14H22O3: C 70.56, H 9.30; found C 70.43, H 9.19.
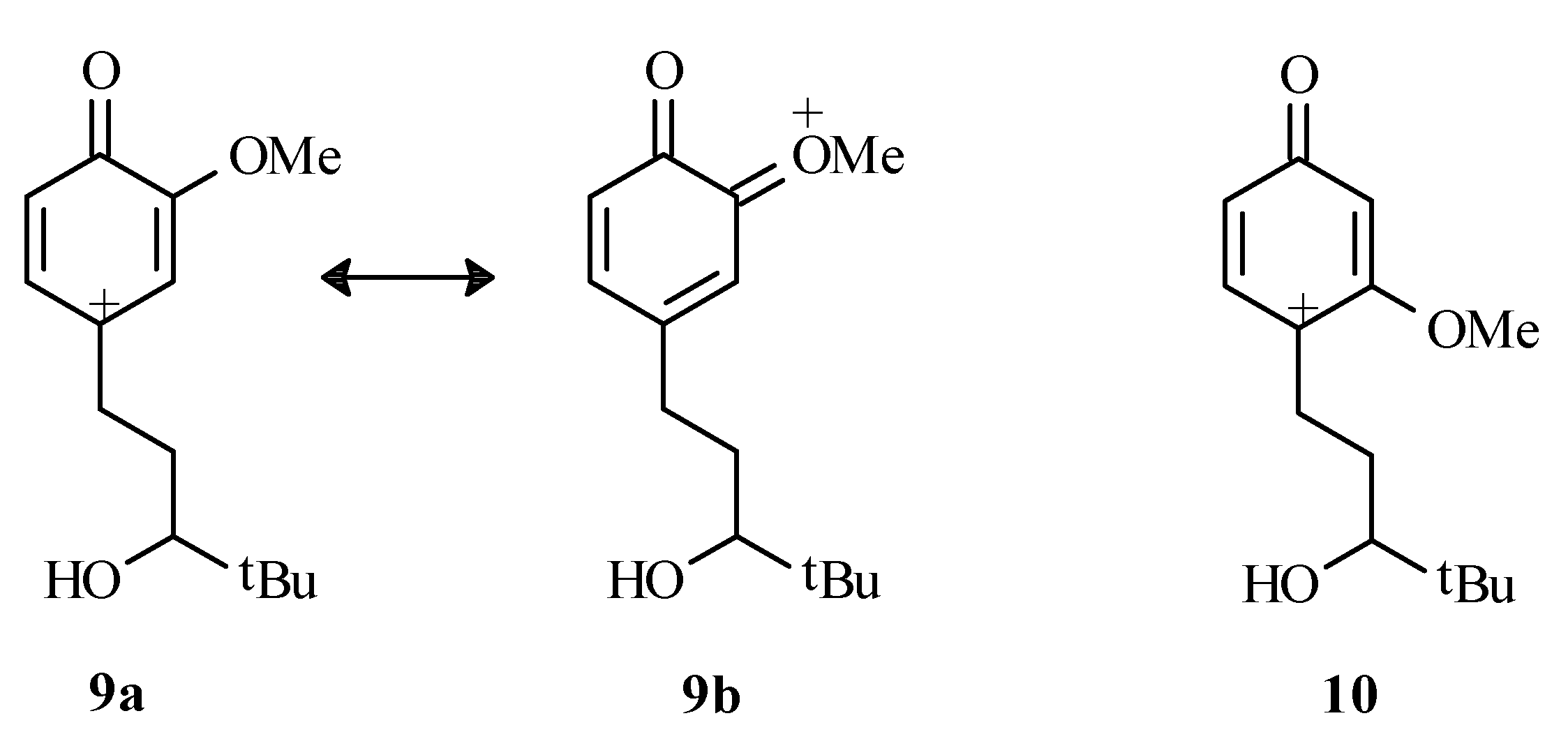
(±)-2-tert-Butyl-6-methoxy-1-oxaspiro[4,5]deca-6,9-diene-8-one (4)
To a cold (0°C) solution of phenol 8 (156 mg, 0.7 mmol) in acetone (20 mL) was added lead tetraacetate (954 mg, 2.2 mmol). The solution was stirred at 0°C for 2 hours, the mixture was filtered through Celite® and ethylene glycol (10 drops) was added. The solution was stirred at room temperature for 20 hours, filtered through Celite® and the solvent was evaporated in vacuo to give a pale yellow oil. 1H-NMR of the crude reaction mixture indicated that two diastereomers were produced in a 50/50 ratio. Chromatography on silica gel (25% EtOAc/hexanes) afforded a pale yellow oil (70 mg, 45%) as a mixture of diastereomers (only one spot by thin layer chromatography). Characterization was performed on the mixture. Whenever distinguishable, values given are for one isomer with those of the second one listed in square brackets. IR (neat) cm-1: 1680 (CO); 1H-NMR (CDCl3) δ: 0.93 [0.95] (s, 9H, t-butyl); 2.10 (m, 5H, H2, H3 and H4), 3.74 [3.77] (s, 3H, OCH3), 5.44 (d, 1H, J=1.7Hz, H7) [5.46 (J=1.7 Hz)], 6.01 (dd, 1H, J=1.7, 9.7Hz, H9) [6.05 (J=1.7, 9.7Hz)], 6.55 (d, 1H, J=9.7, H10) [6.63 (J=9.7Hz)]; 13C-NMR (CDCl3) δ: 26.0 [26.2] (t-butyl CH3), 27.6 [28.1] (C3), 29.8 [33.7] (t-butyl C), 36.8 [37.1] (C4), 55.7 [55.9] (OCH3), 78.2 (C5), 89.5 [91.3] (C2), 101.1 (C7), 126.0 (C9), 147.1 (C10), 175.7 [176.8] (C6), 187.7 (C8); MS m/e (relative %): 236 [M+] (100), 180 (24), 179 (32), 178 (19), 151 (22), 137 (44), 91 (19); Anal. Calc’d for C10H10O4: C 61.81, H 5.19; found C 61.52, H 5.40.

{kind=link}
{kind=link}
{kind=link}


